Descubrimiento y desarrollo de inhibidores de mTOR

Blanco mamífero de rapamicina (mTOR) es un serina/treonina quinasa, que pertenece a fosfatidilinositol 3 quinasa (PI3K) relacionados con la familia de quinasas (PIKKs). Regula la proliferación, el crecimiento y metabolismo celular y por lo tanto es un objetivo para el desarrollo de un número de inhibidores de mTOR.
Efectos vía descendente y formas dos complejos, mTORC1 y mTORC2. Los inhibidores de mTOR más establecidos son llamados rapalogs (rapamicina y sus análogos), que han mostrado las respuestas del tumor en los ensayos clínicos contra varios tipos de tumor.[1]
Contenido
- 1 Historia
- 2 Proteínas quinasas y sus inhibidores
- 3 vía de señalización mTOR
- 3.1 mTOR signaling pathway en cáncer humano
- 4 Desarrollo de inhibidores de mTOR
- 4.1 Inhibidores de mTOR de primera generación
- 4.2 La rapamicina y rapalogs
- 4.2.1 Sirolimus
- 4.2.2 Temsirolimus
- 4.2.3 Everolimus
- 4.2.4 Deforolimus
- 4.3 Inhibidores de mTOR de segunda generación
- 5 Mecanismo de acción
- 5.1 Efectos en las células cancerosas
- 5.2 Efectos sobre la angiogénesis tumoral
- 6 Relación estructura actividad
- 7 Biomarcadores
- 7.1 Sensibilidad
- 8 Inhibidores de la cinasa mTOR ATP-competitivo
- 8.1 inhibidores de mTOR/PI3K duales
- 8.2 inhibidores duales de la mTORC1/mTORC2 (TORCdIs)
- 8.3 Limitaciones de los inhibidores de mTOR de nueva generación
- 9 Futuro
- 10 Véase también
- 11 Referencias
Historia
El descubrimiento de mTOR fue hecho mientras investigaba hace unas décadas la mecanismo de acción de su inhibidor de la, rapamicina.[2][3] La rapamicina fue descubierta en 1975 en una muestra de suelo Isla de Pascua de Pacífico Sur, también conocida como Rapa Nui, de donde se deriva su nombre.[4] La rapamicina es un macrólidos, producida por la microorganismo Streptomyces hygroscopius y mostró antifúngico propiedades. Poco después de su descubrimiento, immuosuppressive las propiedades fueron detectadas, que más tarde condujo a la creación de la rapamicina como un inmunosupresor. En la década de 1980, la rapamicina también fue encontrada para tener actividad anticancerosa aunque el mecanismo exacto de acción permaneció desconocido hasta muchos años después.[2][5][6]
En la década de 1990 hubo un cambio dramático en este campo debido a los estudios sobre el mecanismo de acción de la rapamicina y la identificación de la meta de la droga.[4] Se encontró la rapamicina inhibida proliferación celular y progresión del ciclo celular. Investigación en la inhibición de mTOR ha sido una rama creciente en la ciencia y tiene resultados prometedores.[7]
Proteínas quinasas y sus inhibidores
En general, proteínas quinasas se clasifican en dos grandes categorías basadas en su especificidad de sustrato, kinases de proteína tirosina y proteínas quinasas serina/treonina. Quinasas de especificidad dual son subclase de las tirosina quinasas.[8]
mTOR es una quinasa dentro de la familia de fosfatidilinositol 3 quinasa-relacionadas quinasas (PIKKs),[9] que es una familia de proteínas quinasas serina/treonina, con una similitud de secuencia a la familia de quinasas de lípidos, PI3Ks.[8] Estas quinasas tienen diferentes funciones biológicas,[8] Pero son grandes proteínas con estructura de dominio común.[9]
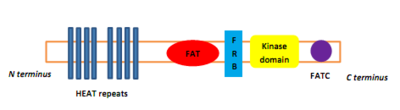
PIKKs tienen cuatro dominios en el nivel de proteína, que los distinguen de otras proteínas quinasas. Desde el N-terminal para el C-terminal, estos dominios son nombrados FRAP-ATM-TRAAP (grasa), el dominio de la cinasa (KD), el dominio PIKK-regulador (PRD) y la grasa-C-terminal (RÖLÓ).[8] El dominio de graso, que consta de cuatro Α-hélices, es N-terminal de KD, sino que parte se conoce como el dominio FKBP12-rapamicina-obligatorio (FRB), que une el complejo FKBP12-rapamicina.[8] El dominio graso consiste en repeticiones, denominadas CALOR (Huntingtina, Factor de elongación 3Una subunidad del proteína fosfatasa 2A y TOR1).[9] Activadores de proteína específica que regulan las quinasas PIKK pero atascamiento de ellos a la quinasa compleja causa un cambio conformacional que aumenta el acceso de sustrato para el dominio de la cinasa.[9]
Kinases de proteína se han convertido en blancos de droga popular.[10] Ellos han sido atacados por el descubrimiento y el diseño de molécula pequeña inhibidores y productos biológicos como posibles agentes terapéuticos. Inhibidores de moléculas pequeñas de proteínas quinasas o evitar generalmente fosforilación de proteínas sustratos o autophosphorylation de la cinasa de sí mismo.[11]
vía de señalización mTOR
Parece ser que factores de crecimiento, los aminoácidos, ATP, y oxígeno los niveles de regulan la señalización mTOR. Varios vías descendentes regulan la progresión del ciclo celular,[12] traducción, iniciación, las respuestas de estrés transcripcional,[13] proteína estabilidad, y supervivencia de las células son señales a través de mTOR.

El serina/treonina quinasa mTOR es un efector aguas abajo de la PI3K/AKT vías y formas dos distintas Complejos Multiproteinas, mTORC1 y mTORC2.[1] Estos dos complejos tienen una red separada de los socios de la proteína, bucles de retroalimentación, sustratosy los reguladores.[14] mTORC1 consiste en dos subunidades reguladoras positivas y mTOR Raptor y mamíferos () LST8mLST8), y dos negativas reguladores, sustrato AKT ricos en prolina 40 (PRAS40) y DEPTOR.[1] mTORC2 se compone de mTOR, mLST8, mSin1, protor, Rictory DEPTOR.[15]
mTORC1 es sensible a la rapamicina pero mTORC2 se considera para ser resistente y es generalmente insensible a señales de energía y nutrientes. mTORC2 se activa por factores de crecimiento, fosforila PKCΑ, AKT y paxilliny regula la actividad de los pequeños GTPasa, RAC, y Rho relacionadas con la supervivencia celular, migración y la regulación de la citoesqueleto de actina.
La cascada de señalización mTORC1 es activada por AKT fosforilada y resultados en la fosforilación de S6K1, y 4EBP1, que conducen a traducción del mRNA.[1]
mTOR signaling pathway en cáncer humano
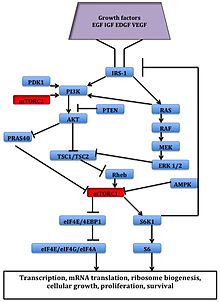
Muchos tumores humanos ocurren debido a una desregulación de señalización mTOR y pueden conferir mayor susceptibilidad a los inhibidores de mTOR.[16] Desregulación de varios elementos de la vía mTOR, como PI3K amplificación/mutación, PTEN pérdida de la función, AKT sobreexpresión y S6K1, 4EBP1, y eIF4E sobreexpresión de haber relacionado con muchos tipos de cánceres. Por lo tanto, mTOR es una interesante Diana terapéutica para el tratamiento de cánceres múltiples, tanto los inhibidores de mTOR ellos mismos o en combinación con inhibidores de otras vías.[1]
Río arriba, señalización PI3K/AKT se desreguló a través de una variedad de mecanismos, incluyendo sobreexpresión o activación de receptores de factor de crecimiento, tales como HER-2 (receptor 2 del factor de crecimiento epidérmico humano) y ANTAGONISTA (receptor del factor de crecimiento insulina-like), mutaciones en PI3K y las mutaciones/amplificaciones de AKT.[1] Homólogo de fosfatasa y Tensina supresor tumoral eliminados en cromosoma diez (PTEN) es un regulador negativo de la señalización PI3K. En muchos cánceres del PTEN expresión disminuye y puede ser regulada a través de varios mecanismos, incluyendo mutaciones, pérdida de heterocigosidad, metilacióny la inestabilidad de la proteína.[15]
Río abajo, la quinasa S6 efectores de mTOR 1 (S6K1), proteína de unión a 4E eucariótico de iniciación factor 1 (4EBP1) y eucariótico de iniciación factor 4E (eIF4E) se relacionan con la transformación celular.[1] S6K1 es un regulador clave del crecimiento celular y también fosforila otros objetivos importantes. Tanto eIF4E y S6K1 están incluidos en transformación celular y su sobreexpresión se ha relacionado con pronóstico pobre cáncer.[15]
Desarrollo de inhibidores de mTOR
Desde el descubrimiento de mTOR, mucha investigación se ha realizado sobre el tema, con rapamicina y rapalogs para entender sus funciones biológicas.[14][17] Los resultados clínicos de dirigidos a esta vía no eran como directo como al principio. Esos resultados han cambiado el curso de la investigación clínica en este campo.[14]
Inicialmente, la rapamicina fue desarrollada como un medicamento antimicótico contra Candida albicans, Aspergillus fumigatus y Cryptococcus neoformans.[5] Pocos años más tarde se detectaron sus propiedades inmunosupresoras. Estudios posteriores condujeron al establecimiento de la rapamicina como un inmunosupresor importante contra rechazo al trasplante, junto con ciclosporina A.[2] Mediante el uso de la rapamicina en combinación con ciclosporina A, mejora la prevención de rechazo en trasplante renal. Por lo tanto, era posible utilizar dosis más bajas de la ciclosporina que reduce al mínimo toxicidad de la droga.[5]
En la década de 1980 la rapamicina fue evaluada por la rama del desarrollo terapéutico del Instituto Nacional del cáncer (NCI). Se descubrió que la rapamicina tuvo una actividad anticancerosa y era un agente no citotóxica con citostático actividad contra varios tipos de cáncer en humanos.[5] Sin embargo, debido a las propiedades farmacocinéticas desfavorables, el desarrollo de inhibidores de mTOR para el tratamiento del cáncer no tuvo éxito en aquel momento.[3] Desde entonces, la rapamicina también ha demostrado ser eficaz para la prevención de la arteria coronaria re-estenosis y para el tratamiento de enfermedades neurodegenerativas.[5]
Inhibidores de mTOR de primera generación
El desarrollo de la rapamicina como agente anticanceroso comenzó otra vez en la década de 1990 con el descubrimiento de temsirolimus (CCI-779). Esto era un derivado de la novela rapamicina soluble que tenía un perfil toxicológico favorable en los animales. Más rapamicina derivados con mejoraron farmacocinética y reducido inmunosupresor efectos desde entonces se han desarrollado para el tratamiento del cáncer.[5] Estos rapalogs son temsirolimus (CCI-779), everolimus (RAD001), y ridaforolimus (AP-23573) que están siendo evaluados en el cáncer ensayos clínicos.[18] La rapamicina análogos tienen efectos terapéuticos similares como rapamicina. Sin embargo han mejorado hidrofilia y puede ser utilizado para oral y administración intravenosa.[4] En 2012 Instituto Nacional del cáncer Listado de más de 200 ensayos clínicos que prueban la actividad anticancerígena de rapalogs tanto como monoterapia con o como parte de terapia de la combinación para muchos tipos de cáncer.[7]
Rapalogs, que son los inhibidores de mTOR de primera generación, han demostrado ser eficaces en un rango de preclínica modelos. Sin embargo, el éxito en ensayos clínicos puede limitarse a sólo unos pocos cánceres raros.[19] Estudios animales y clínicos demuestran que rapalogs son principalmente citostáticosy por lo tanto efectiva como estabilizadores de la enfermedad en lugar de regresión.[20] La tasa de respuesta en tumores sólidos donde rapalogs se han utilizado como agente único terapia han sido modestos. Debido a la inhibición parcial mTOR como se mencionó anteriormente, rapalogs no son suficientes para lograr un efecto anticanceroso amplio y robusto, al menos cuando se utiliza como monoterapia con.[18][19][21]
Otra razón para el éxito limitado es que hay un circuito de retroalimentación entre mTORC1 y AKT en ciertas células tumorales. Parece que la inhibición mTORC1 por rapalogs es incapaz de reprimir un realimentación negativa lazo que se traduce en fosforilación y la activación de AKT.[17][22] Estas limitaciones han llevado al desarrollo de la segunda generación de inhibidores de mTOR.[7]
La rapamicina y rapalogs
La rapamicina y rapalogs (derivados de la rapamicina) son inhibidores de la molécula pequeña,[23] que han sido evaluados como agentes anticancerígenos. Los rapalogs tienen más favorable perfil farmacocinético comparado rapamicina, el fármaco principal,[3] a pesar de los mismos sitios de Unión para mTOR y FKBP12.[5]

Sirolimus
El natural antibiótico, la rapamicina o sirolimus,[6] un agente citostático, ha sido utilizado en terapia de combinación con corticoesteroides y ciclosporina en los pacientes que recibieron trasplante de riñón para evitar rechazo de órganos ambos en los E.e.u.u.[24] y Europa,[25] debido a sus propiedades farmacocinéticas insatisfactorias.[3] En 2003, la U.S. Food and Drug Administration aprobado sirolimus-eluting stents coronarios, que son utilizados en pacientes con estrechamiento de arterias coronarias, o supuestos ateroesclerosis.[26]
Recientemente la rapamicina ha demostrado eficaz en la inhibición del crecimiento de varios cánceres humanos y líneas celulares murinas.[5] La rapamicina es el inhibidor de mTOR principal, pero deforolimus (AP23573), everolimus (RAD001) y temsirolimus (CCI-779), son los análogos de rapamicina desarrollado recientemente.[2]
Temsirolimus
El análogo de la rapamicina, temsirolimus (CCI-779)[2] es también un agente fotoefecto que proliferación tumoral retrasos.
El temsirolimus es pro-droga de la rapamicina. Es aprobado por los Estados Unidos Food and Drug Administration (FDA)[24] y el Agencia Europea de medicamentos (EMA),[27] para el tratamiento del carcinoma de células renales (RCC). Temsirolimus tiene mayor solubilidad en agua que la rapamicina y por lo tanto, es administrado por la inyección intravenosa.[3][6] En 30 de mayo de 2007, fue aprobado por la FDA para el tratamiento del CCR avanzado.[6]
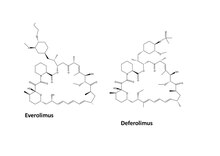
Everolimus
Everolimus es el segunda novela rapamicina análogo.[2] Desde el 30 de marzo de 2009 a 05 de mayo de 2011 la U.S. FDA aprobado para el tratamiento del carcinoma avanzado de células renales después de fracaso del tratamiento con everolimus sunitinib o sorafenib, Astrocitoma de células gigantes subependimario Asociado (SEGA) Esclerosis tuberosa (TS), y Tumores neuroendocrinos progresivos de origen pancreático (PNET).[28] En julio y agosto de 2012, se aprobaron dos nuevas indicaciones, hormona avanzados receptores positivos, HER2-negativo de cáncer de mama en combinación con exemestano y pacientes pediátricos y adultos con SEGA.[28] En 2009 y 2011, también fue aprobado en la Unión Europea para el cáncer de mama avanzado, tumores neuroendocrinos pancreáticos, el carcinoma avanzado de células renales,[29] y SEGA en pacientes con esclerosis tuberosa.[30]
Deforolimus
Deforolimus (AP23573, MK-8669), o ridaforolimus, es la rapamicina nueva análoga y no es un Profármaco.[2] Como el temsirolimus se pueden administrar por vía intravenosa, y está siendo estimado formulación oral para el tratamiento de sarcoma.[3] No fue en mercado en junio de 2012, ya que la FDA quería más humana prueba en él debido a su efectividad y seguridad.[31]
Inhibidores de mTOR de segunda generación
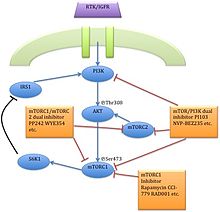
La segunda generación de inhibidores de mTOR es conocida como ATP-competitivo inhibidores de mTOR quinasa.[7] los inhibidores duales de la mTORC1/mTORC2 están diseñados para competir con el ATP en la catalítica sitio de mTOR. Inhiben todas las funciones de la cinasa dependiente de mTORC1 y mTORC2 y por lo tanto, bloquear la activación de retroalimentación de señalización PI3K/AKT, a diferencia de rapalogs que sólo atacan mTORC1.[7][17] Estos tipos de inhibidores han sido desarrollados y varios de ellos se están probando en ensayos clínicos. Como rapalogs, disminuyen la proteína traducción, atenuar ciclo celular la progresión e inhiben angiogénesis en muchas líneas celulares de cáncer y también en el cáncer humano. De hecho han demostrado para ser más potente que la rapalogs.[7]
Teóricamente, las ventajas más importantes de estos inhibidores de mTOR es la considerable disminución de la fosforilación de AKT mTORC2 bloqueo y además de una inhibición mejor en mTORC1.[14] Sin embargo, existen algunos inconvenientes. Aunque estos compuestos han sido eficaces en líneas celulares rapamicina-insensible, solamente han demostrado un éxito limitado en KRAS tumores conducidos. Esto sugiere que combinacional la terapia puede por necesarios para el tratamiento de estos cánceres. Otro inconveniente es también su potencial toxicidad. Estos hechos han expresado su preocupación acerca de la eficacia a largo plazo de estos tipos de inhibidores.[7]
La estrecha interacción de mTOR con la vía PI3K también ha conducido al desarrollo de los inhibidores de mTOR/PI3K duales.[7] En comparación con los fármacos que inhiben la mTORC1 o PI3K, estas drogas tienen el beneficio de la inhibición de mTORC1, mTORC2 y el catalítico isoformas de PI3K. Dirigidos a ambas quinasas al mismo tiempo reduce la upregulation de PI3K, que normalmente se produce con una inhibición en mTORC1.[14] Se ha demostrado la inhibición de la vía PI3K/mTOR potentemente bloquear la proliferación induciendo G1 arresto en líneas celulares tumorales diferentes. Fuerte inducción de apoptosis y autofagia también se ha visto. A pesar de buenos resultados prometedores, hay evidencia preclínica que algunos tipos de cáncer pueden ser insensibles a esta inhibición dual. Los inhibidores duales de PI3K/mTOR también están probables que la toxicidad se han incrementado.[7]
Mecanismo de acción
Los estudios de rapamicina como Agente inmunosupresor nos permitido entender su mecanismo de acción.[5] Inhibe T-cell proliferación y la respuesta proliferativa inducida por varios citoquinas, incluyendo interleuquina 1 (IL-1), IL-2, IL-3, IL-4, IL-6, IGF, PDGF, y factores estimulantes de colonias (CSF).[5] Rapalogs y los inhibidores de rapamicina puede apuntar el crecimiento del tumor tanto directa como indirectamente. Impacto directo de ellos en las células cancerosas dependen de la concentración de la droga y ciertas características celulares. La vía indirecta, se basa en la interacción con los procesos necesarios para el tumor angiogénesis.[5]
Efectos en las células cancerosas
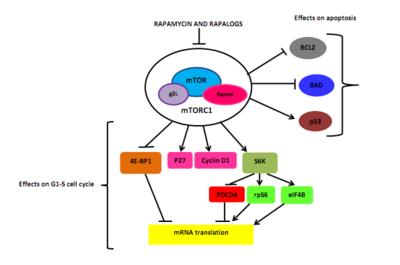
Reticulación rapamicina y rapalogs el Induccin FK506 proteína de Unión tacrolimus o FKBP-12, a través de su Grupo metoxi. El complejo de la rapamicina-FKBP12 interfiere con dominio FRB de mTOR.[5][6] Interacción molecular entre FKBP12, mTOR y rapamicina puede durar unos tres días (72 horas). La inhibición de la mTOR bloquea el atascamiento del raptor accesorio proteína (proteína asociada a reguladores de mTOR) a mTOR, pero que es necesario para aguas abajo fosforilación de la S6K1 y 4EBP1.[5][21]
Como consecuencia, desfosforila S6K1, que reduce la proteína síntesis y disminuye la célula motality y tamaño. La rapamicina induce la desfosforilación de 4EBP1, resultando en un incremento en p27 y una disminución en ciclina D1 expresión. Que conduce a la obstrucción tardía de G1/S ciclo celular. Rapamycin ha demostrado para inducir la muerte celular cáncer estimulando autofagia o apoptosis, pero el mecanismo molecular de la apoptosis en células cancerosas aún no ha sido completamente resuelto. Una sugerencia de la relación entre la inhibición de mTOR y apoptosis puede ser a través de la Diana S6K1, que puede fosforilan MAL, una molécula pro-apoptótica, en Ser136.[5] Esa reacción rompe el atascamiento de malo BCL-XL y BCL2, un mitocondrial inhibidores de la muerte, dando lugar a la inactivación del malo[5] y la supervivencia celular disminuida.[6] Rapamycin también ha mostrado inducir p53-apoptosis independiente en ciertos tipos de cáncer.[5]
Efectos sobre la angiogénesis tumoral
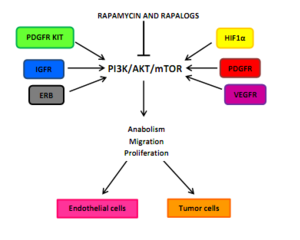
Angiogénesis tumoral dependen de las interacciones entre factores de crecimiento endotelial vasculares todo lo que puede activar el PI3K/AKT/mTOR en las células endoteliales, pericitos, o las células cancerosas. Ejemplo de estos factores de crecimiento son angiopoyetina 1 (ANG1)ANG 2, factor de crecimiento básico del fibroblasto (bFGF), efrina-B2, factor de crecimiento enothelial vascular (VEGF)y los miembros de la factor de crecimiento tumoral-β (TGFβ) Superfamilia. Uno de los principales estímulos de la angiogénesis es la hipoxia, resultando en la activación de factores de transcripción inducible por la hipoxia (HIFs) y la expresión de ANG2, bFGF, PDGF, VEGF y VEGFR. Inhibición de la traducción HIF1α evitando PDGF/PDGFR y VEGF/VEGFR puede resultar de inhibición de mTOR. Un bloqueo del ciclo celular de G0-G1 puede ser la consecuencia de la inactivación de mTOR en células endoteliales y pericitos activado por hipoxia.[5]
Hay algunas pruebas de que la terapia prolongada con rapamicina puede tener efecto sobre AKT y mTORC2 también.[2][32]
Relación estructura actividad
La región pipecolate de la estructura de la rapamicina parece necesaria para enlace de rapamicina a FKBP12. Este paso es necesario para más vinculante de la rapamicina a la mTOR quinasa, que es la enzima clave en muchas de las acciones biológicas de la rapamicina.[33]
La alta afinidad de unión de rapamicina a FKBP12 se explica por el número de enlaces de hidrógeno a través de dos diferentes hidrofóbico vinculante los bolsillos y esto ha sido revelado por la estructura cristalina radiografía del compuesto a la proteína. Las características estructurales comunes de temsirolimus y sirolimus; el ácido pipecólico, región tricarbonyl de C13-C15, y lactona funcionalidades del papel clave en grupos con la FKBP12 vinculantes.[18][34]
El más importante enlaces de hidrógeno son el oxígeno carbonilo lactona en C-21 al columna vertebral NH de Ile56, carbonilo de la amida en C-15 al grupo fenólico de la cadena lateral de Tyr82y el protón del hidroxilo en el hemiketal carbono, C-13, en el circuito sidechain de Asp37.[34]
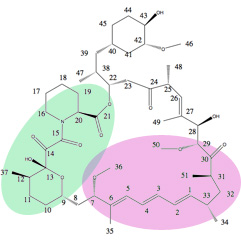
Los cambios estructurales a la estructura de la rapamicina pueden afectar vinculante a mTOR. Esto podría incluir vinculante tanto directo como indirecto como parte de la Unión a FKBP12. Interacción del complejo FKBP12-rapamicina con mTOR corresponde con flexibilidad conformacional del dominio efector de la rapamicina. Este dominio se compone de las regiones moleculares que hacen interacciones hidrofóbicas con el dominio FKB y Triene región de C-1-C-6, grupo metoxilo en C-7, y grupos metilo C-33, C-27 y C-25. Todos los cambios del anillo macrólido pueden tener efectos imprevisibles en vinculantes y por lo tanto, hacer determinación de SAR para rapalogs problemático.[34][35]
La rapamicina no contiene grupos funcionales que ionizar En pH rango 1-10 y por lo tanto, son bastante insolubles en agua.[23] A pesar de su eficacia en modelos de cáncer preclinic, su pobre solubilidad en el agua, la estabilidad y la eliminación de vida media larga dificultaba su uso parenteral, pero el desarrollo de análogos de la rapamicina soluble había vencido varias barreras.[2]
No obstante, los análogos de la rapamicina que han sido aprobados para el uso humano se modifican en el grupo hidroxilo C-43 y muestran una mejoría en farmacocinética parámetros, así como propiedades de droga, por ejemplo la solubilidad.[35]
La rapamicina y temsirolimus tienen estructuras químicas similares y se unen a FKBP12, aunque su mecanismo de acción difiere.[18]
El temsirolimus es un dihydroxymethyl propiónico ácido Ester la rapamicina, y su primera derivada.[2] Por lo tanto es más agua soluble y debido a su watersolubility puede darse mediante la formulación intravenosa.[6][18]
Everolimus O-2 hidroxietil sustitución de cadena y deforolimus tiene un óxido de fosfina substitución en la posición C-43 en el anillo de lactona de la rapamicina.[18]
Deforolimus (Ridaforolimus) tiene C43 moiety alcohol secundario del grupo de la rapamicina que fue sustituido con fosfonato cyclohexyl y grupos fosfinato, evitando la alta afinidad de unión a mTOR y FKBP. Estudios computacionales de modelización ayudó a la synthesise del compuesto.[6]
Biomarcadores
Identificación de predictivo biomarcadores de eficacia para tipos de tumores que son sensibles a los inhibidores de mTOR sigue siendo un grave problema.[1][36] Posibles biomarcadores predictivos para respuesta tumoral a los inhibidores de mTOR, como se han descrito en glioblastoma, Mama y cáncer de próstata las células, pueden ser la expresión diferencial de proteínas vía mTOR, PTEN, AKTy S6.[1] Por lo tanto, esta información se basa en los ensayos preclínicos, basados en in vitro líneas celulares tumorales, que sugieren que los efectos de los inhibidores de mTOR pueden ser más pronunciados en los cánceres mostrando pérdida de las funciones PTEN cultivadas o PIK3CA mutaciones. Sin embargo, el uso de PTEN, PIK3CA mutaciones, y AKT – phospho estatus para predecir la sensibilidad rapalog no se ha validado totalmente en clínica. Hasta la fecha, intenta identificar biomarcadores de rapalog respuesta ha tenido éxito.[20]
Sensibilidad
Clínicos y traslacionales datos indican que tipos de tumor sensible, con los parámetros adecuados y funcionales apoptosis las vías, no necesitarán altas dosis de inhibidores de mTOR para desencadenar la apoptosis. En la mayoría de los casos, las células cancerosas pueden ser parcialmente sensibles a los inhibidores de mTOR debido a redundante transducción de señales o la falta de apoptosis funcional rutas de señalización. En situaciones como ésta, pueden ser necesarias altas dosis de inhibidores de mTOR. En un estudio reciente de pacientes con Carcinoma de células renales, resistencia al Temsirolimus se asoció con bajos niveles de p-AKT y p-S6K1, que juegan el papel clave en la activación de mTOR. Estos datos sugieren fuertemente número de tumores con una vía de señalización PI3K/AKT/mTOR activado que no responde a los inhibidores de mTOR. Para futuros estudios se recomienda para excluir a los pacientes con niveles bajos o negativos p-AKT de ensayos con inhibidores de mTOR.
Los datos actuales son insuficientes para predecir la sensibilidad de los tumores de la rapamicina. Sin embargo, los datos existentes nos permiten caracterizar los tumores que no pueden responder a rapalogs.[5]
Inhibidores de la cinasa mTOR ATP-competitivo
Estos inhibidores de mTOR enlazar al sitio de unión a ATP en dominio de la cinasa mTOR requerido para las funciones de ambos de segunda generación mTORC1 y mTORC2y resultar en downregulation de mTOR vía de señalización. Debido a la capacidad de PI3K y mTORC2 para regular la fosforilación de AKT, estos dos compuestos desempeñan un papel clave para minimizar la activación de retroalimentación de AKT.[19]
inhibidores de mTOR/PI3K duales
Varios, llamados mTOR/PI3K duales inhibidores (TPdIs), se han desarrollado y están en etapa temprana ensayos preclínicos y muestran resultados prometedores. Su desarrollo ha sido beneficiado con estudios previos con los inhibidores selectivos de PI3K.[19] La actividad de estas moléculas pequeñas de rapalog actividad difiere de la manera mediante el bloqueo de ambos phospholylation mTORC1 dependiente de S6K1 y mTORC2-dependiente de la fosforilación de AKT Ser473 residuo.[1]

Inhibidores de mTOR/PI3K duales incluyen NVP-BEZ235, BGT226, SF1126, PKI-587 y muchos más. Por ejemplo, Novartis ha desarrollado el compuesto NVPBE235 que se informó para inhibir el crecimiento tumoral en modelos preclínicos. Mejora la actividad antitumoral de algunas otras drogas tales como vincristina.[19] NVP-BEZ235 parece inhibir eficazmente tanto tipo salvaje y mutante forma de PIK3CA, que sugiere su uso hacia amplio tipos de tumores. Los estudios han demostrado la actividad antiproliferativa superior a rapalogs y en vivo modelos han confirmado estos potentes antineoplásicos efectos de los inhibidores de mTOR/PI3K duales.[1][7] Objetivo de estos inhibidores isoformas de PK3I (p110α, β y γ) junto con sitios de unión a ATP de mTORC1 y mTORC2 mediante el bloqueo de señalización PI3K/AKT, incluso en los tipos de cáncer con mutaciones en esta vía.[7]
inhibidores duales de la mTORC1/mTORC2 (TORCdIs)

Nuevos inhibidores de mTOR específicos salieron de proyección y descubrimiento de fármacos esfuerzos. Estos compuestos bloquean la actividad de ambos complejos de mTOR y se denominan inhibidores duales de mTORC1/mTORC2.[19] Compuestos con esta características tales como INK128, AZD8055 y AZD2014 han entrado ensayos clínicos. Se han estudiado una serie de estos inhibidores de mTOR quinasa. Su estructura se deriva Morfolino pyrazolopyrimidine andamio.[19][21] Han realizado mejoras de este tipo de inhibidores mediante el intercambio de las morfolinas con morfolinas puenteado en inhibidores de la pyrazolopyrimidine y los resultados mostraron mayor selectividad a mTOR por doblez 26000.[21][37]
Limitaciones de los inhibidores de mTOR de nueva generación
Aunque la nueva generación de inhibidores de mTOR gran promesa para la terapia contra el cáncer y se está moviendo rápidamente en ensayos clínicos, hay muchas cuestiones importantes que determinan su éxito en la clínica. Primero de todo predecibles biomarcadores para el beneficio de estos inhibidores no están disponibles. Parece que los determinantes genéticos predisponen a las células cancerosas sensibles o resistentes a estos compuestos. Los tumores que dependen de la vía PI3K/mTOR deben responder a estos agentes, pero no está claro si los compuestos son eficaces en los cánceres con distintas lesiones genéticas.[19]
Inhibición de mTOR es una estrategia prometedora para el tratamiento del número de cánceres. Actividad clínica limitada de agentes selectivos mTORC1 han hecho poco probable que tengan impacto en el tratamiento del cáncer. El desarrollo de inhibidores de la ATP-catalítico compatitive tienen la capacidad de bloquear la mTORC1 y el mTORC2.[38]
Futuro
Las limitaciones de rapalogs disponibles actualmente han llevado a nuevos enfoques dirigidos a mTOR. Los estudios sugieren que los inhibidores de mTOR pueden tener actividad anticancerosa en muchos tipos de cáncer, tales como RCC, tumores neuroendocrinos, cáncer de mama, carcinoma hepatocelular, sarcoma, y linfoma de células B grandes.[3] Una limitación importante para el desarrollo de la terapia de inhibición de mTOR es que no están actualmente disponibles para predecir qué paciente responderá a los biomarcadores. Una mejor comprensión de los mecanismos moleculares que intervienen en la respuesta de las células cancerosas a los inhibidores de mTOR se requiere para que esto sea posible.[7]
Puede ser una manera de vencer la resistencia y mejorar la eficacia de mTOR dirigidos a agentes con estratificación de los pacientes y selección de terapias de combinación de drogas. Esto puede conducir a una terapia anticáncer más efectiva y personalizada.[1][7] Aunque se necesitan más investigaciones, mTOR targering sigue siendo una opción terapéutica atractiva y prometedora para el tratamiento del cáncer.[7]
Véase también
- Blanco mamífero de rapamicina (mTOR)
- Vía PI3K/AKT/mTOR
- Vía de señalización Akt/PKB
- Inhibidor de PI3K
Referencias
- ^ a b c d e f g h i j k l Pópulo, Helena; Lopes, José Manuel; Soares, Paula (2012). "La vía de señalización en cáncer humano mTOR". Revista Internacional de Ciencias moleculares 13 (12): 1886 – 918. Doi:10.3390/ijms13021886. PMC3291999. PMID22408430.
- ^ a b c d e f g h i j Strimpakos, Alex S.; Karapanagiotou, Eleni M.; Saif, Wasif M.; Syrigos, Kostas N. (2009). "El papel de mTOR en el tratamiento de tumores sólidos: una descripción". Comentarios sobre tratamiento de cáncer 35 (2): 148 – 59. Doi:10.1016/j.CTRV.2008.09.006. PMID19013721.
- ^ a b c d e f g Yuan, Ruirong; Kay, Andrea; Berg, William J; Lebwohl, David (2009). "Targeting tumorigénesis: desarrollo y uso de inhibidores de mTOR en terapia del cáncer". Diario de Hematología y Oncología 2:: 45. Doi:10.1186/1756-8722-2-45. PMC2775749. PMID19860903.
- ^ a b c Tsang, Chi Kwan; Qi, Haiyan; Liu, Leroy F.; Zheng, X.F. Steven (2007). "Objetivos de blanco mamífero de rapamicina (mTOR) para la salud y las enfermedades". Hoy el descubrimiento de fármacos 12 (3 – 4): 112 – 24. Doi:10.1016/j.Drudis.2006.12.008. PMID17275731.
- ^ a b c d e f g h i j k l m n o p q r Faivre, Sandrine; Kroemer, Guido; Raymond, Eric (2006). "Desarrollo actual de los inhibidores de mTOR como agentes anticancerígenos". Descubrimiento de fármacos de naturaleza comentarios 5 (8): 671 – 88. Doi:10.1038/nrd2062. PMID16883305.
- ^ a b c d e f g h VIGNOT, S.; Faivre, S; Aguirre, D; Raymond, E (2005). "Terapia orientada a MTOR de cáncer con rapamicina derivados". Anales de Oncología 16 (4): 525 – 37. Doi:10.1093/annonc/mdi113. PMID15728109.
- ^ a b c d e f g h i j k l m n Zaytseva, Yekaterina Y.; Valentino, Joseph D.; Gulhati, Pat; Evers, B. (2012). "Inhibidores de MTOR en terapia del cáncer". Letras de cáncer 319 (1): 1 – 7. Doi:10.1016/j.canlet.2012.01.005. PMID22261336.
- ^ a b c d e Lempiäinen, Harri; Halazonetis, Thanos D (2009). "Emerging temas comunes en el Reglamento de PIKKs y PI3Ks". La revista EMBO 28 (20): 3067 – 73. Doi:10.1038/emboj.2009.281. PMC2752028. PMID19779456.
- ^ a b c d Lovejoy, Courtney A.; Cortez, David (2009). "Los mecanismos comunes de regulación PIKK". Reparación del ADN 8 (9): 1004 – 8. Doi:10.1016/j.dnarep.2009.04.006. PMC2725225. PMID19464237.
- ^ McConnell, J. L.; Wadzinski, B. E. (2009). "Objetivos de serina/treonina proteína fosfatasas para el desarrollo de fármacos". Farmacología molecular 75 (6): 1249 – 61. Doi:10.1124/mol.108.053140. PMC2684880. PMID19299564.
- ^ Grant, S. K. (2008). "Inhibidores de la cinasa de proteína terapéutica". Ciencias de la vida celular y Molecular 66 (7): 1163 – 77. Doi:10.1007/s00018-008-8539-7. PMID19011754.
- ^ https://www.ncbi.nlm.nih.gov/Books/NBK9962/[completa citación necesitada]
- ^ Divida, esteras (2007). "La respuesta de estrés de transcripción". Ciclo celular 6 (18): 2252 – 7. Doi:10.4161/CC.6.18.4751. PMID17700065.
- ^ a b c d e Vilar, E.; Perez-Garcia, J.; Tabernero, J. (2011). "Empujar la envoltura en la vía mTOR: la segunda generación de inhibidores". Molecular oncológica 10 (3): 395-403. Doi:10.1158/1535-7163.MCT-10-0905. PMC3413411. PMID21216931.
- ^ a b c Meric-Bernstam, f el.; Gonzalez-Angulo, A. M. (2009). "Objetivos de la red de señalización mTOR para el tratamiento del cáncer". Journal of Clinical Oncology 27 (13): 2278 – 87. Doi:10.1200/JCO.2008.20.0766. PMC2738634. PMID19332717.
- ^ Huang, S; Houghton, PJ (2003). "Objetivos de mTOR de señalización para el tratamiento del cáncer". Current Opinion in farmacología 3 (4): 371 – 7. Doi:10.1016/S1471-4892 (03) 00071-7. PMID12901945.
- ^ a b c Ballou, Lisa M.; Lin, Richard Z. (2008). "Inhibidores de la cinasa de la rapamicina y mTOR". Revista de Biología química 1 (1 – 4): 27 – 36. Doi:10.1007/s12154-008-0003-5. PMC2698317. PMID19568796.
- ^ a b c d e f Brachmann, Saskia; Fritsch, Christine; Maira, Saveur-Michel; García-Echeverría, Carlos (2009). "Inhibidores de PI3K y mTOR — una nueva generación de agentes anticancerosos dirigidos". Current Opinion in biología celular 21 (2): 194 – 8. Doi:10.1016/j.CEB.2008.12.011. PMID19201591.
- ^ a b c d e f g h Zhang, Yan-Jie; Duan, Yanwen; Zheng, Steven X.F. (2011). "Contra el dominio de la cinasa mTOR: la segunda generación de inhibidores de mTOR". Hoy el descubrimiento de fármacos 16 (7 – 8): 325 – 31. Doi:10.1016/j.Drudis.2011.02.008. PMC3073023. PMID21333749.
- ^ a b Vagabundeo, Seth A.; Hennessy, Bryan T.; Slingerland, Joyce M. (2011). "Inhibidores de mTOR de próxima generación en oncología clínica: Cómo complejidad vía informa estrategia terapéutica". Revista de investigación clínica 121 (4): 1231 – 41. Doi:10.1172/JCI44145. PMC3069769. PMID21490404.
- ^ a b c d Tanneeru, Karunakar; Guruprasad, Lalitha (2011). Generación pharmacophore 3D basado en ligando y acoplamiento molecular de inhibidores de mTOR quinasa. Diario de Modelado Molecular 18 (4): 1611 – 24. Doi:10.1007/s00894-011-1184-3. PMID21805127.
- ^ Sutherlin, Daniel P.; Bao, Linda; Berry, Megan; Castanedo, Georgette; Chuckowree, Irina; Dotson, Jenna; Amigos, Adrian; Friedman, Lori; Goldsmith, Richard; Gunzner, Janet; Heffron, Timothy; Lesnick, John; Lewis, Cristina; Mathieu, Simon; Murray, Jeremy; Nonomiya, Jim; Pang, Jodie; Pegg, Niel; Prior, Wei Wei; Rouge, Lionel; Salphati, Laurent; Sampath, Deepak; Tian, Qingping; Tsui, Vickie; WAN, Nan Chi; Wang, Shumei; Wei, Binqing; Wiesmann, cristiano; Wu, Ping; Zhu, Bing-Yan (2011). "Descubrimiento de un potente y selectivo oral disponible clase I fosfatidilinositol 3-quinasa (PI3K) / mamíferos objetivo de rapamicina (mTOR) inhibidor de la cinasa (GDC-0980) para el tratamiento del cáncer". Journal of Medicinal Chemistry 54 (21): 7579 – 87. Doi:10.1021/jm2009327. PMID21981714.
- ^ a b Simamora, Pahala; Alvarez, Joan M; Yalkowsky, Samuel H (2001). "Solubilización de la rapamicina". Revista Internacional de farmacia 213 (1 – 2): 25 – 9. Doi:10.1016/s0378-5173 (00) 00617-7. PMID11165091.
- ^ a b "Libro naranja: aprobado medicamentos con evaluaciones de equivalencia terapéutica". Food and Drug Administration. 25 de septiembre de 2012.
- ^ "Rapamune". Agencia Europea de medicamentos. 25 de septiembre de 2012.
- ^ "CYPHER ™ Sirolimus-eluting Stent coronario - P020026". Food and Drug Administration. 25 de septiembre de 2012.
- ^ "Torisel". Agencia Europea de medicamentos. 25 de septiembre de 2012.
- ^ a b "Aprobación de la FDA para Everolimus". Instituto Nacional del cáncer. 20 de septiembre de 2012.
- ^ "Afinitor". Agencia Europea de medicamentos. 25 de septiembre de 2012.
- ^ "Votubia". Agencia Europea de medicamentos. 25 de septiembre de 2012.
- ^ "La FDA quiere más pruebas Ridaforolimus". Desarrollo y descubrimiento de fármacos. 20 de septiembre de 2012.
- ^ Garcia‑Echeverria, Carlos (2011). "Bloqueando la vía mTOR: una perspectiva de descubrimiento de droga". Transacciones de la sociedad bioquímica 39 (2): 451 – 5. Doi:10.1042/BST0390451. PMID21428918.
- ^ Ritacco, F. V.; Graziani, E. I.; Los veranos, M. Y.; Zabriskie, T. M.; Yu, K.; Bernan, V. S.; Carter, G. T.; Greenstein, M. (2005). "Producción de análogos de la rapamicina novela por biosíntesis Precursor-dirigida". Microbiología Aplicada y ambiental 71 (4): 1971 – 6. Doi:10.1128/AEM.71.4.1971-1976.2005. PMC1082568. PMID15812028.
- ^ a b c Abraham, Robert T.; Gibbons, James J.; Graziani, Edmund I. (2010). "Química y Farmacología de la rapamicina y sus derivados". En el Hall, Michael N.; Tamanoi, Fuyuhiko. Estructura, función y regulación de TOR complejos de levaduras a los mamíferos. Las enzimas 27. págs. 329 – 66. Doi:10.1016/S1874-6047 (10) 27017-8. ISBN978-0-12-381539-2.
- ^ a b Barrish, Joel C.; Carter, Percy; Cheng, Peter et al., eds (2010). Las cuentas en el descubrimiento de fármacos: estudios de caso en química Medicinal. Cambridge: Real Sociedad de química. ISBN978-1-84973-126-3. [Página necesitado]
- ^ Delbaldo, Catherine; Albert, Sébastien; Dreyer, Chantal; Sablin, Marie-Paule; Serova, Maria; Raymond, Eric; Faivre, Sandrine (2011). "Biomarcadores predictivos para la actividad de la Diana de los inhibidores de rapamicina (mTOR) mamíferos". Oncología dirigida 6 (2): 119 – 24. Doi:10.1007/s11523-011-0177-6. PMID21533544.
- ^ Nowak, Pawel; Cole, Derek C.; Brooijmans, Natasja; Bursavich, Matthew G.; Curran, Kevin J.; Ellingboe, John W.; Gibbons, James J.; Hollander, Irwin; Hu, Yongbo; Kaplan, Josué; Malwitz, David J.; Toral-Barza, Lourdes; Verheijen, Jeroen C.; Zask, Arie; Zhang, Wei-Guo; Yu, Ker (2009). "El descubrimiento de potente y los inhibidores selectivos de la blanco mamífero de rapamicina (mTOR) Kinase". Journal of Medicinal Chemistry 52 (22): 7081 – 9. Doi:10.1021/jm9012642. PMID19848404.
- ^ Altman, Jessica K.; Sassano, Antonella; Platanias, Leonidas C. (2011-06-14). "Objetivos de mTOR para el tratamiento de esta afección. Nuevos agentes y nuevas direcciones". Oncotarget 2 (6): 510-517. PMC3248202. PMID21680954.
|
||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Otras Páginas
- Fantastic Voyage: Vivir lo suficiente para vivir por siempre
- Comparacion de servicios de copia de seguridad en linea
- Banca de inversion (seccion estructura organizativa)
- Sistema de gestion de flujo de datos
- Los cerveceros del arroz
- Eutanasia de insectos
- Teatro de Dryden
- Establecimiento estructurado (seccion ventas de estructurado liquidacion pagos despues de estructurado establecimiento se ha establecido)
- Ingresos no operacionales
- Softline International
- Destilar redes