Hiperplasia suprarrenal congénita
| Hiperplasia suprarrenal congénita | |
|---|---|
| Clasificación y recursos externos | |
| CIE-10 | E25.0 |
| CIE-9 | 255.2 |
| OMIM | 201910 201710 202110 201810 202010 |
| DiseasesDB | 1854 1832 4 1841 2565 |
| MedlinePlus | 000411 |
| eMedicine | PED/48 |
| Malla | D000312 |
Hiperplasia suprarrenal congénita (CAH) son cualquiera de varios autosómico recesivo ligado al sexo enfermedades resultantes de mutaciones de genes para enzimas mediando los pasos bioquímicos de la producción de cortisol De colesterol por la glándulas suprarrenales (esteroidogénesis).[1]
La mayoría de estas condiciones implica la producción excesiva o deficiente de esteroides sexuales y puede alterar el desarrollo de primaria o características secundarias del sexo en algunos niños afectados, niños o adultos.[2]
Contenido
- 1 Signos y síntomas
- 2 Genética
- 2.1 Penetrancia
- 3 Diagnóstico
- 3.1 Evaluación clínica
- 3.2 Estudios de laboratorio
- 3.3 Clasificación
- 4 Proyección
- 5 Tratamiento
- 6 Epidemiología
- 7 Historia
- 7.1 Antes del siglo XX
- 7.2 siglo XX y XXI
- 8 Véase también
- 9 Referencias
- 10 Lectura adicional
- 11 Enlaces externos
Signos y síntomas
Los síntomas de la CAH varían dependiendo de la forma de CAH y el sexo del paciente. Los síntomas pueden incluir:
Debido a la inadecuada mineralocorticoides:
- vómitos debido a pérdida de sal conduce a deshidratación y la muerte
Debido a los andrógenos exceso:
- funcional y promedio de tamaño pene en casos de extrema virilización (pero sin esperma)
- órganos genitales ambiguos, en algunas hembras, así que puede ser inicialmente difícil identificar los órganos genitales externos como "hombre" o "mujer".
- temprano vello púbico y un rápido crecimiento en la infancia
- pubertad precoz o el fracaso de pubertad que se produzca)Infantilismo sexual:: ausente o retraso de la pubertad)
- excesivo vello facial, virilización, o Irregularidad menstrual en la adolescencia
- infertilidad debido a anovulación
- agrandado clítoris y superficial vagina[3]
Genética
El gen de 21-hidroxilasa se encuentra en 6p21.3 como parte de la HLA complejo. deficiencia de 21-hidroxilasa da lugar a una mutación única con dos altamente homólogas cerca-copias en serie que consta de un gen activo (CYP21A) y un pseudogene inactivo (CYP21P). Los alelos mutantes resultan de recombinación entre los genes activos y pseudo (conversión del gene).[4]
Penetrancia
Mayor variabilidad es introducida por el grado de enzima ineficiencia producida por el específico alelos cada paciente tiene. Algunos alelos como resultado grados más severos de la ineficiencia de la enzima. En general, severos grados de ineficiencia producen cambios en el feto y problemas en la vida prenatal o perinatal. Grados más leves de ineficiencia están generalmente asociados con excesiva o deficiente hormonas sexuales efectos en la niñez o la adolescencia, mientras que la forma más leve de CAH interfiere con la ovulación y fertilidad en los adultos.
Diagnóstico
Evaluación clínica
Los bebés femeninos con CAH clásica tienen genitales ambiguos debido a la exposición a altas concentraciones de andrógenos en el útero. CAH debido a la deficiencia de 21-hidroxilasa es la causa más frecuente de genitales ambiguos en los bebés femeninos genotípicamente normales (46XX). Menos mujeres severamente afectadas pueden presentar con Pubarquia temprana. Las mujeres jóvenes pueden presentar con síntomas de síndrome de ovario poliquístico (oligomenorrea, ovarios poliquísticos, hirsutismo).
Los machos con CAH clásica generalmente no tienen ningún signo de CAH al nacer. Algunos pueden presentar hiperpigmentación y posible ampliación del pene. Edad de diagnóstico de varones con CAH varía y depende de la severidad de la deficiencia de aldosterona. Niños con pérdida de sal enfermedad presente temprano con síntomas de hiponatremia y la hipovolemia. Chicos con sal-perder enfermedad presente después con signos de virilización.[4]
Estudios de laboratorio
En el clásico 21-hidroxilasa, estudios de laboratorio mostrará concentraciones muy altas de 17-hidroxiprogesterona (más de 242 nmol/L en la muestra de sangre al azar; con la normal es de menos de 3 nmol/L en la edad de 3 días en un bebé a término). Sal-perder pacientes tienden a tener niveles más altos de 17-hidroxiprogesterona que los pacientes no-sal-perder.
Resultados falsos positivos de la investigación neonatal para CAH pueden verse en los recién nacidos prematuros. Muchos programas de cribado tienen referencia específica varía dependiendo de la edad gestacional y peso.
En casos dudosos acerca de CAH, puede realizarse una prueba de estimulación de corticotropina.
El análisis genético puede ser útil para confirmar un diagnóstico de CAH, pero no es necesario si existen clásico clínico y los resultados del laboratorio.
Clasificación
El cortisol es un suprarrenal hormona esteroidea Es necesaria para la normal función endocrina. La producción comienza en el segundo mes de vida fetal. Producción de cortisol pobre es un rasgo distintivo de la mayoría de las formas de CAH. Resultados de la producción de cortisol ineficientes en aumento de los niveles de ACTH, que a su vez induce crecimiento excesivo ()hiperplasia) y la hiperactividad de la esteroide-producción de las células de la corteza suprarrenal. Los defectos que causan hiperplasia suprarrenal son congénita (es decir, presente al nacer).
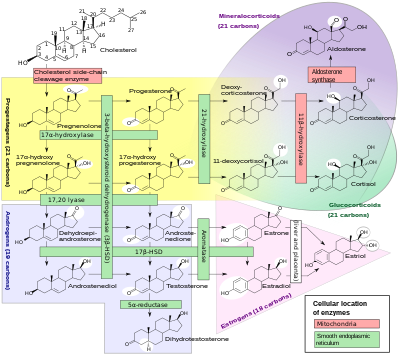
Deficiencia del cortisol en CAH suele ser parcial y no el problema más grave para una persona afectada. Síntesis del cortisol comparte pasos con la síntesis de mineralocorticoides tales como aldosterona, andrógenos tales como testosterona, y estrógenos tales como estradiol. La producción excesiva o deficiente resultante de estas tres clases de hormonas producen los problemas más importantes para las personas con CAH. Ineficiencias de enzimas específicas se asocian a patrones característicos de excesiva o insuficiente de los mineralocorticoides o esteroides sexuales.
| Término médico común | % | OMIM | Enzyme(s) | Lugar geométrico | Substratos | Producto (s) | Mineralocorticoides | Andrógenos |
|---|---|---|---|---|---|---|---|---|
| CAH 21-hidroxilasa | 90-95% | 201910 | P450c21 | 6p21.3 | 17OH-progesterona→ progesterona→ |
11-deoxycortisol DOC |
↓ | ↑ |
| CAH 11β-hidroxilasa | 5% | 202010 | P450c11β | 8q21-22 | 11-deoxycortisol→ DOC→ |
cortisol corticosterona |
↑ | ↑ |
| CAH 3Β-HSD | muy raro | 201810 | 3ΒHSD II | 1 p 13 | pregnenolona→ 17OH-pregnenolona→ DHEA→ |
progesterona 17OH-progesterona androstenediona |
↓ | ↓ |
| CAH 17α-hidroxilasa | muy raro | 202110 | P450c17 | 10q24.3 | pregnenolona→ progesterona→ 17OH-pregnenolone→ |
17OH-pregnenolona 17OH-progesterona DHEA |
↑ | ↓ |
| lipoidea CAH (20,22-desmolase) |
muy raro | 201710 | Estrella P450scc |
8p11.2 15q23-q24 |
transporte de colesterol colesterol→ |
en las mitocondrias pregnenolona |
↓ | ↓ |
Desde los años sesenta que la mayoría endocrinólogos se han referido a las formas de CAH por los nombres en la columna de la izquierda tradicionales, que generalmente corresponden a la actividad de la enzima deficiente. Como estructuras exactas y los genes de las enzimas se identificaron en la década de 1980, la mayoría de las enzimas fueron encontrada para ser oxidasas del citocromo P450 y fueron retitulados para reflejar esto. En algunos casos, más de una enzima fue encontrada para participar en una reacción, y en otros casos una sola enzima mediada en más de una reacción. También hubo variación en diferentes tejidos y especies de mamíferos.
En todas sus formas, hiperplasia suprarrenal congénita debido a la deficiencia de 21-hidroxilasa representa aproximadamente el 95% de los casos diagnosticados de CAH. A menos que otra enzima específica se menciona, se nombra "CAH" en casi todos los contextos 21-hidroxilasa deficiencia. (Los términos "pérdida de sal CAH", y "CAH virilizante simple" generalmente se refiere a los subtipos de esta condición.) CAH debido a deficiencias de enzimas distintos de 21-hidroxilasa presentan muchos de los mismos desafíos de gestión como la deficiencia de 21-hidroxilasa, pero algunas implican mineralocorticoide exceso o esteroides sexuales deficiencia.
Proyección
En la actualidad, en los Estados Unidos y más de 40 países, cada recién nacido es defendido para CAH en el nacimiento. Esta prueba detecta los niveles elevados de 17-hidroxi-progesterona (17-OHP). Detectar niveles altos de 17-OH progesterona permite la detección temprana de CAH. Los recién nacidos detectan a tiempo suficiente puede colocarse en la medicación y vivir una vida relativamente normal.
Sin embargo, el proceso de selección, se caracteriza por un alto índice de falsos positivos. En un estudio,[5] Proyección de CAH tuvo la menor Valor predictivo positivo (111 casos verdaderos positivos entre 20.647 anormal proyección resultados en un período de dos años, o 0,53%, compararon con 6,36% por deficiencia de biotinidasa, 1.84% para congénita hipo-tiroidismo, 0,56% para galactosemia clásica y 2,9% para la fenilcetonuria). Según esta estimación, 200 recién nacidos afectados requieren clínica y seguimiento de laboratorio para cada caso de CAH.
Tratamiento
El tratamiento de todas las formas de CAH puede incluir cualquiera de:
- abastecimiento suficiente glucocorticoide para reducir la hiperplasia y la sobreproducción de andrógenos o mineralocorticoides
- proporcionando mineralocorticoide reemplazo y extra sal si la persona es deficiente
- prestación de reemplazo testosterona o estrógeno en la pubertad si la persona es deficiente
- tratamientos adicionales para optimizar el crecimiento por retrasar la pubertad o retrasar maduración ósea
Todos estos temas de gestión se discuten con más detalle en hiperplasia suprarrenal congénita debido a la deficiencia de 21-hidroxilasa.
Dexametasona se utiliza como una fuera de etiqueta el tratamiento prenatal temprano para los síntomas de la CAH en los fetos femeninos, pero no trata el trastorno subyacente congénito. Un ensayo clínico sueco 2007 encontró que tratamiento puede provocar defectos cognitivos y conductuales, pero el pequeño número de sujetos de prueba significa que el estudio no puede considerarse definitivo. Un estudio americano 2012 encontró más positivo que los resultados adversos.[6] La administración de dexametasona prenatal ha sido objeto de controversia sobre cuestiones de consentimiento informado y porque el tratamiento debe anteriores a un diagnóstico clínico de CAH en el feto femenino.[7] Sobre todo porque en el útero dexametasona puede causar problemas metabólicos que no son evidentes hasta más adelante en vida.[citación necesitada]
El tratamiento también ha generado preocupaciones en la LGBT Comunidad tras un ensayo publicado en el foro de la Hastings Center, un think-tank dedicado a Bioética, que citó investigaciones publicadas que sugirió que tratamiento prenatal de fetos femeninos podría evitar que los fetos se lesbianas después del nacimiento, puede hacerlas más propensas a participar en carreras y comportamiento "tradicionalmente" mujer identificada y más interesado en rodamientos y criar a los hijos. Citando un intento conocido por un hombre usando su conocimiento de la efecto de la orden fraternal de nacimiento para evitar tener una homosexual hijo mediante el uso de un sustituto, los ensayistas (profesor Alice Dreger de Northwestern University Feinberg School of Medicine, profesora Ellen Feder de American University y abogada Anne Tamar-Mattis) sugieren que los tratamientos pre-natal "dex" constituyen el primer intento conocido por utilizar en el útero protocolos para reducir la incidencia de la homosexualidad y bisexualidad en los seres humanos.[8] Encuentran tal manipulación para ser moralmente censurable.
CAH es un gen recesivo, la madre y del padre deben ser recesivos portadores de CAH para un niño tener CAH. Debido a los avances en la medicina moderna, esas parejas con los genes recesivos de CAH tienen una opción para prevenir la CAH en su descendencia a través de diagnóstico genético preimplantacional (DGP). En PGD, el óvulo es fecundado fuera de cuerpo de la mujer en una placa Petri (FIV). En el tercer día, cuando el embrión ha desarrollado a partir de una célula a aproximadamente 4 a 6 células, una de esas células es removida del embrión sin dañar el embrión. El embrión sigue creciendo hasta el día 5 cuando está congelada o implantado en la madre. Mientras tanto, la célula quitada es analizada para determinar si el embrión tiene CAH. Si el embrión está decidido a tener CAH, los padres pueden tomar una decisión en cuanto a si desean tenerlo implantados en la madre o no.[citación necesitada]
Metanálisis de los estudios apoyan el uso de dexametasona en fetos en riesgo CAH encontró "menos de la mitad del uno por ciento de los estudios publicados de esta intervención fueron mirados como de alta calidad suficiente para proporcionar datos significativos para un metanálisis. Incluso estos cuatro estudios eran de baja calidad". "de manera tan descuidada en cuanto a incumplimiento de las normas profesionales de ética médica"[9] y "no había ningún dato sobre el seguimiento a largo plazo de los resultados físicos y metabólicos en los niños expuestos a la dexametasona".[10]
Epidemiología
La incidencia varía geográficamente. En los Estados Unidos, la hiperplasia suprarrenal congénita es particularmente común en nativos americanos y esquimales (incidencia de Yupic 1⁄280). Entre los americanos caucásicos, la incidencia es de aproximadamente 1⁄15.000).[4]
Historia
Antes del siglo XX
Anatomista italiano, Luigi De Crecchio proporciona la descripción más temprana conocida de un caso de probable CAH.
Propongo en esta narrativa que a veces es extremadamente difícil e incluso imposible de determinar sexo durante la vida. En uno de los anatómica teatros del hospital..., llegó hacia finales de enero un cadáver que en la vida era el cuerpo de un cierto Joseph Marzo... La fisonomía general era decididamente masculina en todos los aspectos. No hubo ningún curvas femeninas en el cuerpo. Hubo una gran barba. Hubo algún manjar de estructura con los músculos que no fueron muy bien desarrollados. La distribución de vello púbico era típico del macho. Tal vez las extremidades más bajas fueron un poco delicadas, que se asemeja a la hembra y estaban cubiertas de pelo... El pene Posteriormente fue curvado y mide 6 cm, o con estiramientos, 10 cm. de la corona era 3 cm de largo y 8 cm de circunferencia. Hubo una amplia prepucio. Hubo un primer grado Hipospadias... Hubo dos pliegues de piel procedentes de la parte superior del pene y lo rodea a ambos lados. Éstos eran un poco flojo y asemejado majora de las labias.
De Crecchio describió los órganos internos, que incluyó un normal vagina, útero, tubos, y ovarios.
Era de gran importancia para determinar los hábitos, tendencias, las pasiones y carácter general de esta persona... Estaba decidido a conseguir como completar una historia posible, decidida a llegar a la base de los hechos y para evitar la excesiva exageración que proliferaba en la conversación de muchas de las personas presentes en el momento de la disección.
Entrevistó a muchas personas y él mismo satisfecho que Joseph Marzo "llevado a cabo él mismo dentro del área sexual exclusivamente como un varón", incluso hasta el punto de contratar al "Enfermedad francesa"en dos ocasiones. La causa de muerte fue otro en una serie de episodios de vómitos y diarrea.[11]
Este relato fue traducido por Alfred Bongiovanni de Crecchio ("Sopra un caso di apparenzi virili en una donna". Morgagni 7:154 – 188, 1865) en 1963 por un artículo en El New England Journal of Medicine.
siglo XX y XXI
La Asociación de esteroides efectos excesivo sexo con enfermedades de la corteza suprarrenal ha sido reconocida por más de un siglo. El término síndrome adrenogenital se aplicó a ambos tumores producir esteroides sexuales y formas severas de CAH durante gran parte del siglo XX, antes de que algunas de las formas de CAH eran entendidos. La hiperplasia suprarrenal congénita, que también data de la primera mitad del siglo, se ha convertido en el término preferido para reducir la ambigüedad y acentuar la patofisiología subyacente de los trastornos.
Mucha de nuestra comprensión moderna y el tratamiento de CAH proviene de investigaciones realizadas en Johns Hopkins Medical School en Baltimore a mediados del siglo XX. Lawson Wilkins, "fundador" de Endocrinología pediátrica, funcionó la patofisiología aparentemente paradójica: que la hiperplasia y la superproducción de andrógenos suprarrenales resultaron de deteriorada capacidad para la fabricación de cortisol. Denunció la utilización de extractos corticales suprarrenales para tratar a niños con CAH en 1950. Cirugía reconstructiva genital también fue pionero en Hopkins. Después de la aplicación de Cariotipo CAH y otro Intersexualidad trastornos en la década de 1950, John MoneyHampson JL y JG Hampson persuadió a la comunidad científica y el público que asignación del sexo no debería basarse en cualquier criterio biológico único, y la identidad de género en gran parte fue aprendida y no tiene ninguna relación sencilla con cromosomas o las hormonas. Ver Intersexualidad para una historia completa, incluyendo las recientes controversias sobre la cirugía reconstructiva.
Hidrocortisona, fludrocortisona, y prednisona estaban disponibles en la década de 1950. En 1980 todos los esteroides pertinentes podrían medirse en la sangre por los laboratorios de referencia para el cuidado del paciente. En 1990 habían sido identificadas casi todos los genes específicos y enzimas.
Sin embargo, la última década ha visto un número de nuevos desarrollos, discutido más ampliamente en hiperplasia suprarrenal congénita debido a la deficiencia de 21-hidroxilasa:
- debate sobre el valor de cirugía reconstructiva genital y cambiar las normas
- debate sobre asignación del sexo de recién nacidos gravemente virilizados de XX
- nuevos tratamientos para mejorar los resultados de altura
- evaluación del recién nacido programas para detectar CAH al nacer
- aumento de intentos de tratar antes del nacimiento de la CAH
Véase también
- Hiperplasia suprarrenal congénita debido a la deficiencia de 21-hidroxilasa
- Hiperplasia suprarrenal congénita debido a 3-hidroxiesteroide deshidrogenasa
- Hiperplasia suprarrenal congénita debido a la 11 beta-hidroxilasa
- Hiperplasia suprarrenal congénita debido a la deficiencia de 17 alfa-hidroxilasa
- Trastornos del desarrollo sexual
- Errores congénitos del metabolismo esteroideo
Referencias
- ^ Warrell (2005). Manual Oxford de medicina: secciones 18-33. Oxford University Press. págs. 261 –. ISBN978-0-19-856978-7. 14 de junio de 2010.
- ^ Aubrey Milunsky; Jeff Milunsky (29 de enero de 2010). Trastornos genéticos y el feto: diagnóstico, prevención y tratamiento. Juan Wiley e hijos. PP. 600 –. ISBN978-1-4051-9087-9. 14 de junio de 2010.
- ^ Richard D. McAnulty, M. Michele Burnette (2006) Sexo y la sexualidad, volumen 1, Greenwood Publishing Group, p.165
- ^ a b c Mais, Daniel D. (2008). Rápido compendio de patología clínica (2ª ed.). Chicago: Prensa ASCP. ISBN0891895671.
- ^ Kenneth A. Pass, Eurico Camargo Neto (2005). Actualización: Evaluación del recién nacido para endocrinopatías. págs. 831 – 834. 12 de diciembre de 2013.
- ^ Meyer-Bahlberg, H. "cognitivo resultado de descendencia de los embarazos tratados con dexametasona en riesgo de hiperplasia suprarrenal congénita debido a la deficiencia de 21-hidroxilasa." European Journal of Endocrinology. Julio de 2012, 167
- ^ Elton, Catherine (2010-06-18). "Un tratamiento Prenatal plantea cuestiones de ética médica". TIEMPO. 2010-07-05.
- ^ Dreger, Alice; Ellen K. Feder; Anne Tamar-Mattis (2010-06-29). "Prevención de la homosexualidad (y arrogante mujeres) en el útero".. Foro de Bioética, un servicio del centro Hastings. 2010-07-05.
- ^ Dreger, Alice; Ellen K. Feder; Anne Tamar-Mattis (30 de julio de 2012). "Dexamethasone prenatal para la hiperplasia suprarrenal congénita". Revista de investigación Bioética 9 (3): 277-294. Doi:10.1007/s11673-012-9384-9. PMC3416978. PMID22904609.
- ^ Fernández-Balsells, M.M.; K. Muthusamy; G. Smushkin; et al (2010). "Uso de dexametasona prenatal para la prevención de virilización en embarazos de riesgo para la hiperplasia suprarrenal congénita clásica debido a deficiencia de 21-hidroxilasa (CYP21A2): una revisión sistemática y meta-análisis". Clinical Endocrinology 73 (4): 436 – 444. Doi:10.1111/j.1365-2265.2010.03826.x. PMID20550539.
- ^ Bongiovanni, Alfred M.; Raíz, Allen W. (1963). "El Síndrome Adrenogenital". El New England Journal of Medicine 268 (23): 1283. Doi:10.1056/NEJM196306062682308. PMID13968788.
Lectura adicional
- Han, Thang S.; Walker, Brian R.; Arlt, Wiebke; Ross, Richard J. (17 de diciembre de 2013). "Resultados de salud y tratamiento en adultos con hiperplasia suprarrenal congénita". Naturaleza comentarios Endocrinología 10 (2): 115-124. Doi:10.1038/nrendo.2013.239. PMID24342885Figura 2: El camino de la esteroidogénesis suprarrenal.
Enlaces externos
| Wikimedia Commons tiene medios relacionados con Hiperplasia suprarrenal congénita. |
- Hiperplasia suprarrenal congénita en DMOZ
- Hijo: Hiperplasia suprarrenal congénita Sistema de salud de la Universidad de Michigan
- Historia de Anre
- Presentación de Gorsuch, y (08 de julio de 2011)"Trastornos suprarrenales en la infancia y la adolescencia"
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||