Síndrome de cáncer

A síndrome de cáncer o síndrome de cáncer familiar es un trastorno genético en el cual heredó mutaciones genéticas en uno o más genes predisponen a los individuos afectados al desarrollo de cáncer y también puede causar el inicio temprano de estos cánceres. Síndromes de cáncer a menudo muestran no sólo un alto riesgo de por vida de desarrollar cáncer, sino también el desarrollo de múltiples tumores primarios independientes.[1] Muchos de estos síndromes son causados por mutaciones en genes supresores de tumores, los genes que están involucrados en la protección de la célula se conviertan en cancerosas. Otros genes que podrían ser afectadas son Reparación del ADN genes, oncogenes y los genes implicados en la producción de los vasos sanguíneos (angiogénesis).[2] Son ejemplos comunes de síndromes de cáncer hereditario síndrome de cáncer de mama-ovario hereditario y cáncer de colon hereditario sin poliposis (Síndrome de Lynch).[3][4]
Contenido
- 1 Fondo
- 2 Genética
- 3 Algunos ejemplos
- 3.1 Anemia de Fanconi
- 3.2 Poliposis adenomatosa familiar
- 3.3 Hereditario cáncer de mama y ovario
- 3.4 Cáncer de colon hereditario sin poliposis
- 3.5 Síndrome hereditario paraganglioma-feocromocitoma
- 3.6 Síndrome de Li-Fraumeni
- 3.7 Poliposis asociada a MUTYH
- 3.8 Síndrome del carcinoma basocelular nevoide
- 3.9 Enfermedad de von Hippel-Lindau
- 3.10 Xerodermia pigmentosa
- 4 Referencias
Fondo
Síndromes de cáncer hereditario subyacen en 5 a 10% de todos los cánceres.[5] La comprensión científica de los síndromes de susceptibilidad de cáncer está expandiendo activamente: síndromes adicionales se han encontrado,[5] la biología subyacente es cada vez más clara, y comercialización de la metodología de diagnóstico genética está mejorando el acceso clínico.[citación necesitada] Dada la prevalencia de cáncer de mama y de colon, la mayoría ampliamente reconocido síndromes incluyen síndrome de cáncer de mama-ovario hereditario (HBOC) y cáncer de colon hereditario sin poliposis (HNPCC, síndrome de Lynch).[5]
Algunos cánceres raros están fuertemente asociadas con síndromes de cáncer hereditario predisposición. Las pruebas genéticas se deben considerar con carcinoma corticosuprarrenal; tumores carcinoides; difuso cáncer gástrico; trompa de Falopio, primario cáncer peritoneal; Leiomiosarcoma; cáncer medular de tiroides; paraganglioma/Pheochromocytoma; carcinoma de células renales de cromófobo, oncocítica híbrido, o oncocitoma histología; carcinoma sebáceo; y los tumores de la médula con túbulos anulares del sexo.[5]
Genética

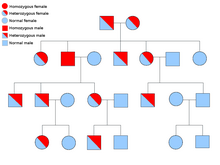
Dos copias de cada gen están presentes en todas las células del cuerpo y cada uno es llamado un alelo. La mayoría de síndromes de cáncer son transmitidos en un mendeliana autosómica dominante manera. En estos casos, sólo un alelo defectuoso tiene que estar presente para una persona tener una predisposición al cáncer. Los individuos con un alelo normal y un alelo defectuoso son conocidos como heterocigoto. Un individuo heterocigoto y una persona con dos alelos normales (homocigótico) tendrá un 50% de posibilidades de producir un niño afectado.[6] La mutación en el gen hereditario es conocida como un mutación de línea germinal y una mutación en el alelo normal más resultados en el desarrollo del cáncer. Esto se conoce como dos hipótesis golpe de Knudson, donde el primer golpe del gen es la mutación hereditaria y el segundo golpe ocurre más adelante en la vida.[2] Como sólo un alelo necesita ser transformado (en comparación con ambos en el llamado "cánceres esporádicos"), el individuo tiene una mayor probabilidad de desarrollar el cáncer que la población general.[citación necesitada]
Con menos frecuencia, síndromes pueden transmitirse como un autosómica recesiva rasgo. Ambos alelos de un gen deben ser transformados en trastornos autosómicos recesivos que un individuo tiene una predisposición al cáncer. Una persona con dos alelos recesivos se conoce como homocigótica recesiva . Ambos padres deben tener al menos un alelo defectuoso para un niño ser homocigótica recesiva. Si ambos padres tienen un alelo mutante y un alelo normal (heterocigoto) entonces tienen un 25% de probabilidad de producir un niño homocigótico recesivo (tiene predisposición), 50% de posibilidades de producir un niño heterozigótico (portador del gen defectuoso) y 25% de probabilidades de produce un niño con dos alelos normales.[6]
Son ejemplos de síndromes de cáncer autosómica dominante Síndrome linfoproliferativo autoinmune (Síndrome de Canale-Smith) Síndrome de Beckwith-Wiedemann (aunque el 85% de los casos son esporádico),[citación necesitada] Síndrome de Birt-Hogg-Dubé, Síndrome de Carney, familiar Cordoma, Síndrome de Cowden, síndrome nevo displástico con melanoma familiar, poliposis adenomatosa familiar, síndrome de cáncer de mama-ovario hereditario, hereditario difuso cáncer gástrico (DHCG), cáncer de colon hereditario sin poliposis (Síndrome de Lynch) Síndrome de Howel-Evans de cáncer eosophageal con tylosis, síndrome de poliposis juvenil, Síndrome de Li-Fraumeni, neoplasia endocrina múltiple tipo 1/2, Osteocondromatosis múltiple, neurofibromatosis tipo 1/2, síndrome del carcinoma basocelular nevoide (Síndrome de Gorlin) Síndrome de Peutz-Jeghers, familiar cáncer de próstata, leiomiomatosis hereditaria cáncer de células renales (LRCC), hereditaria papilar cáncer de células renales (HPRCC), hereditaria paraganglioma-síndrome de feocromocitoma retinoblastoma, esclerosis tuberosa, enfermedad de von Hippel-Lindau y Tumor de Wilms.[7]
Son ejemplos de síndromes de cáncer recesivo autosómico ataxia-telangiectasia, Síndrome de Bloom, Anemia de FanconiPoliposis asociada a MUTYH, Síndrome de Rothmund-Thomson, Síndrome de Werner y Xerodermia pigmentosa.[7]
Algunos ejemplos
Aunque síndromes de cáncer presentan un mayor riesgo de cáncer, el riesgo varía. Para algunas de estas enfermedades, el cáncer no es su principal característica. La discusión se centra en su asociación con un mayor riesgo de cáncer. Esta lista dista de ser exhaustiva.
Anemia de Fanconi
Anemia de Fanconi (FA) es un desorden con un espectro clínico amplio, incluyendo: inicio temprano y mayor riesgo de cáncer; insuficiencia de la médula ósea; y anomalías congénitas. Las manifestaciones más importantes de este trastorno son las relacionadas a hematopoeisis (producción de sangre por la médula ósea); Estos incluyen anemia aplásica, síndrome mielodisplásico y leucemia mieloide aguda. Tumores hepáticos y carcinomas de células escamosas de la esófago, orofaringe y úvula son tumores sólidos comúnmente asociados con FA. Anomalías congénitas incluyen: anomalías esqueléticas (especialmente aquellas que afectan a las manos), manchas café au lait y hipopigmentación. Hasta la fecha, los genes causan FA son: FANCA, FANCB, FANCC, FANCD2, FANCE, FANCF, FANCG, FANCI, FANCJ, FANCL, FANCM, FANCN, FANCO, FANCP y BRCA2 (anteriormente conocido como FANCD1). La herencia de este síndrome es principalmente autosómica recesiva, pero FANCB puede ser heredada de la maternal o paternal cromosoma x (herencia recesiva ligado al cromosoma x). La vía FA está implicada en la DNA reparación cuando las dos hebras de ADN son incorrectamente unieron ()reticulaciones intercatenarios). Muchos caminos son coordinados por la vía de FA para esta incluida reparación de la supresión del nucleótido, síntesis de translesion y recombinación homóloga.[8][9][10][11][12]
Poliposis adenomatosa familiar
Poliposis adenomatosa familiar (FAP) es una autosómica dominante síndrome que aumenta considerablemente el riesgo de cáncer colorrectal. Aproximadamente 1 de cada 8000 personas tendrá esta enfermedad y tiene aproximadamente 100% penetrancia. Una persona con esta enfermedad tendrá cientos o miles de benigno adenomas a lo largo de su Colón, que va en progreso más casos de cáncer. Otros tumores aumentados en frecuencia incluyen; osteomas, suprarrenal adenomas y carcinomas, tumores de la tiroides y tumores desmoides. La causa de este desorden es una mutación Gen APC, que está implicado en Β-catenina regulación. APC defectuoso causa β-catenina se acumulan en las células y activar factores de transcripción involucrado en proliferación de células, migración, diferenciación y apoptosis (muerte celular programada).[13][14][15]
Hereditario cáncer de mama y ovario
Síndrome de cáncer de mama-ovario hereditario (HBOC) es un autosómica dominante trastorno genético causada por mutaciones genéticas de la BRCA1 y BRCA2 genes. En las mujeres este trastorno principalmente aumenta el riesgo de Mama y cáncer de ovario, pero también aumenta el riesgo de carcinoma de la trompa de Falopio y carcinoma seroso papilar del peritoneo. En hombres el riesgo de cáncer de próstata se incrementa. Otros tipos de cáncer que están sistemáticamente vinculadas a este síndrome son cáncer pancreático, cáncer de mama masculino, cáncer colorrectal y los cánceres de la útero y cuello uterino. Las mutaciones genéticas representan aproximadamente el 7% y 14% del cáncer de mama y ovario, respectivamente y representan el 80% de estos casos BRCA1 y BRCA2. BRCA1 y BRCA2 son ambos genes supresores de tumores implicado en el mantenimiento y reparación de ADN. Las mutaciones en estos genes permiten mayores daños al ADN, que puede conducir al cáncer.[16][17]
Cáncer de colon hereditario sin poliposis
Cáncer de colon hereditario sin poliposis (HNPCC), también conocido como síndrome de Lynch, es una autosómica dominante síndrome de cáncer que aumenta el riesgo de cáncer colorrectal. Es causada por mutaciones genéticas en Reparación del ADN desajuste Los genes (MMR), en particular MLH1, MSH2, MSH6 y PMS2. Además de cáncer colorrectal muchos otros tipos de cáncer se incrementan en frecuencia. Estos incluyen; cáncer de endometrio, cáncer de estómago, cáncer de ovario, cáncer del intestino y cáncer pancreático. HNPCC también está asociado con un inicio temprano de cáncer colorrectal. Los genes MMR están involucrados en la reparación de ADN cuando el bases en cada hebra de ADN no coinciden. Los genes MMR defectuosos permiten continuos inserción y eliminación mutaciones en las regiones de ADN conocidas como microsatélites. Estos breves secuencias repetitivas de ADN inestable, llevando a un estado de inestabilidad de microsatélites (MSI). Microsatélites mutados suelen encontrarse en los genes implicados en la progresión y la iniciación tumoral, y MSI puede mejorar la supervivencia de las células, conduce al cáncer.[4][18][19][20]
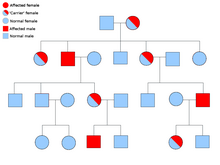
Síndrome hereditario paraganglioma-feocromocitoma
Mayoría de los casos del paraganglioma familiar es causada por mutaciones en el Succinato deshidrogenasa (SDH; oxidorreductasa succinato: Ubiquinona) (genes de la subunidadSDHD, SDHAF2, SDHC, SDHB).
PGL-1 se asocia con la mutación de SDHD y PGL-1 mayoría de los individuos con paraganglioma ha afectado a los padres en lugar de madres afectadas. PGL1 y PGL2 son autosómico dominante con impresión. PGL-4 se asocia SDHB mutación y se asocia con un mayor riesgo de feocromocitoma, así como cáncer de células renales y cáncer de tiroides medular-no.[21]
Síndrome de Li-Fraumeni
Síndrome de Li-Fraumeni es un autosómica dominante síndrome causado principalmente por mutaciones En Gene TP53, que aumenta el riesgo de muchos cánceres grandemente y también se asocia altamente a la aparición temprana de estos cánceres. Los cánceres ligados a este trastorno incluyen; sarcomas de partes blandas (a menudo se encuentran en la infancia), osteosarcoma, cáncer de mama, cáncer de cerebro, leucemia y carcinoma corticosuprarrenal. Los individuos con el síndrome de Li-Fraumeni suelen tienen múltiples cánceres primarios independientes. La razón para el amplio espectro clínico de este trastorno puede deberse a otras mutaciones genéticas que modifican la enfermedad. La proteína producida por el gene TP53, p53, está involucrada en detención del ciclo celular, Reparación del ADN y apoptosis. P53 defectuoso no puede ser capaz de realizar correctamente estos procesos, que puede ser la razón para la formación del tumor. Porque sólo 60-80% de las personas con este trastorno tienen mutaciones detectables en TP53, otras mutaciones en la vía de p53 pueden implicar en el síndrome de Li-Fraumeni.[22][23][24][25]
Poliposis asociada a MUTYH
Poliposis asociada MUTYH comparte la mayoría de sus características clínicas con FAP; la diferencia es que es un autosómica recesiva desorden causado por mutaciones en el MUTYH Reparación del ADN Gene. Los tumores con mayor riesgo en este desorden son cáncer colorrectal, los adenomas gástricos y duodenales adenomas.[13][26]

Síndrome del carcinoma basocelular nevoide
Síndrome del carcinoma basocelular nevoide (NBCCS), también conocido como síndrome de Gorlin, es un autosómica dominante síndrome de cáncer en el cual el riesgo de carcinoma de células basales es muy alta. La enfermedad se caracteriza por basocelular nevos, mandíbula queratoquistes y anormalidades esqueléticas. Las estimaciones de prevalencia NBCCS varía, pero es aproximadamente 1 en 60000. La presencia de carcinoma de células basales es mucho mayor en blanco que individuos negros; 80% y 38%, respectivamente. Queratoquistes odontogénicos se encuentran en aproximadamente el 75% de las personas con la enfermedad y a menudo ocurre temprano en la vida. Las anormalidades esqueléticas más frecuentes ocurren en la cabeza y la cara, pero otras áreas son a menudo afectadas tales como la caja torácica. El causativo mutación genética de esta enfermedad se presenta en el Gene de PTCH, y el producto de PTCH es un supresor de tumores involucrado en señalización celular. Aunque se desconoce la función exacta de esta proteína en NBCCS, está implicado en la vía de señalización de erizo, conocido por control crecimiento de la célula y el desarrollo.[27][28]
Enfermedad de von Hippel-Lindau
Enfermedad de von Hippel-Lindau Enfermedad (VHL) es una condición genética dominante rara, de un autosoma que predispone a las personas con tumores benignos y malignos. Los tumores más comunes en la BVS son el sistema nervioso central y hemangioblastomas retinianos, carcinomas renal de células claras, los feocromocitomas, tumores neuroendocrinos pancreáticos, quistes pancreáticos, tumores del saco endolinfático y cistoadenomas papilar del epidídimo.[29][30] BVS resulta de una mutación en el gen supresor tumoral de von Hippel-Lindau en el cromosoma 3p25.3.[31]
Xerodermia pigmentosa
Xerodermia pigmentosa (XP) es una autosómica recesiva trastorno que se caracteriza por una sensibilidad a luz ultravioleta (UV), masivamente un mayor riesgo de quemaduras de sol y mayor riesgo de cánceres de piel. El riesgo de cáncer de piel es más de 10000 veces de individuos normales e incluye muchos tipos de cáncer de piel, incluyendo melanoma y cánceres de piel no melanoma. Además, sol expuesto áreas de la lengua, los labios y ojos tienen un mayor riesgo de convertirse en cancerosos. XP puede asociarse con otros cánceres internos y tumores benignos.[citación necesitada] Además de cáncer, algunos mutaciones genéticas esa causa XP están asociados con neurodegeneración. XP puede ser causado por mutaciones genéticas en 8 genes que producen los siguientes enzimas: XPA, XPB, XPC, XPD, XPE, XPF, XGP y Pol η. Son XPA-XPF reparación de la supresión del nucleótido enzimas que reparacion UV luz-dañaron el ADN y las proteínas defectuosas permitirá la acumulación de mutaciones causadas por la luz UV. Pol η es un polimerasa, que es una enzima implicada en la replicación del ADN. Hay muchos polimerasas, pero pol η es la enzima que se replica el ADN dañado por la luz UV. Las mutaciones en este gene produce una enzima η pol defectuosa que no puede replicar el ADN con daño de luz ultravioleta. Los individuos con mutaciones de este gen tienen un subconjunto de XP; XP-variante de la enfermedad.[32][33]
Referencias
- ^ Simone Fulda; Heike Allgayer; Helga Rehder (2009). Los tumores hereditarios: Desde los Genes a consecuencias clínicas. Weinheim: Wiley-VCH. ISBN3-527-32028-8.
- ^ a b Hodgson S (enero de 2008). "Mecanismos de susceptibilidad al cáncer heredado". J Zhejiang Univ Sci B 9 (1): 1 – 4. Doi:10.1631/jzus.B073001. PMC2170461. PMID18196605.
- ^ Según Clark, Domchek SM (abril de 2011). "Manejo clínico de síndromes de cáncer de mama hereditario". J la glándula mamaria Biol Neoplasia 16 (1): 17 – 25. Doi:10.1007/s10911-011-9200-x. PMID21360002.
- ^ a b Lynch HT, Lynch PM, Lanspa SJ, Snyder CL, Lynch JF, Boland CR (julio de 2009). "Informe sobre el síndrome de Lynch: historia, genética molecular, detección, diagnóstico diferencial y ramificaciones médico-legales". Clin. Genet. 76 (1): 1 – 18. Doi:10.1111/j.1399-0004.2009.01230.x. PMC2846640. PMID19659756.
- ^ a b c d Los bancos KC, Moline JJ, Marvin ML, Newlin AC, Vogel KJ. 10 los tumores raros que garantizan una derivación genética. Fam cáncer. 2013 mar; 1:1-18. doi: 10.1007/s10689-012-9584-9. PMID 23377869
- ^ a b Anderson, Cindy Lou; Carie A Braun (2007). Fisiopatología: alteraciones funcionales en la salud humana. Hagerstwon, MD: Lippincott Williams & Wilkins. ISBN0-7817-6250-2.
- ^ a b Lindor NM, Greene MH (julio de 1998). "El manual conciso de síndromes de cáncer familiar. Programa de cáncer familiar de mayo". J. nacional Cancer Inst. 90 (14): 1039 – 71. Doi:10.1093/jnci/90.14.1039. PMID9672254.
- ^ Moldovan GL, d ' Andrea AD (2009). "Cómo la vía de la anemia de fanconi guardas del genoma". Annu. El reverendo Genet. 43:: 223 – 49. Doi:10.1146/annurev-jineta-102108-134222. PMC2830711. PMID19686080.
- ^ Tischkowitz MD, Hodgson SV (enero de 2003). "La anemia de Fanconi". Journal of Medical Genetics 40 (1): 1 – 10. Doi:10.1136/JMG.40.1.1. PMC1735271. PMID12525534.
- ^ Kee Y, Andrea AD (noviembre de 2012). "Manejo clínico de la anemia de Fanconi y patogenia molecular". J. Clin. Invertir. 122 (11): 3799 – 806. Doi:10.1172/JCI58321. PMC3484428. PMID23114602.
- ^ Kottemann MC, Smogorzewska (enero de 2013). La anemia de Fanconi y la reparación de Watson y Crick ADN vínculos cruzados. Naturaleza 493 (7432): 356 – 63. Doi:10.1038/nature11863. PMID23325218.
- ^ Su X, Huang J (septiembre de 2011). "El camino de la anemia de Fanconi y ADN interstrand reticulación reparación". Proteína celular 2 (9): 704 – 11. Doi:10.1007/s13238-011-1098-y. PMID21948210.
- ^ a b Media E, D Bercovich, Rozen P (2009). "Poliposis adenomatosa familiar". Orphanet J rara Dis 4:: 22. Doi:10.1186/1750-1172-4-22. PMC2772987. PMID19822006.
- ^ Galiatsatos P, Foulkes WD (febrero de 2006). "Poliposis adenomatosa familiar". AM j Gastroenterol. 101 (2): 385 – 98. Doi:10.1111/j.1572-0241.2006.00375.x. PMID16454848.
- ^ MacRae F, du Sart D, Nasioulas S (2009). "Poliposis adenomatosa familiar". Best Pract Res Clin Gastroenterol 23 (2): 197-207. Doi:10.1016/j.BPG.2009.02.010. PMID19414146.
- ^ N Petrucelli, Daly MB, Feldman GL (mayo de 2010). Mama hereditario y el cáncer de ovario debido a las mutaciones en el BRCA1 y BRCA2. Genet. Med. 12 (5): 245 – 59. Doi:10.1097/GIM.0b013e3181d38f2f. PMID20216074.
- ^ Smith EC (2012). "Tener una visión general de mama hereditario y el síndrome de cáncer de ovario". J Midwifery Womens Health 57 (6): 577 – 84. Doi:10.1111/j.1542-2011.2012.00199.x. PMID23050669.
- ^ Drescher KM, Sharma P, Lynch HT (2010). "Las hipótesis actuales sobre cómo inestabilidad de microsatélites conduce a mayor supervivencia de los pacientes con síndrome de Lynch". Clin. Dev. Immunol. 2010:: 170432. Doi:10.1155/2010/170432. PMC2901607. PMID20631828.
- ^ Kunkel TA, Erie DA (2005). "La reparación del ADN desajuste". Annu. El reverendo Biochem. 74:: 681 – 710. Doi:10.1146/annurev.Biochem.74.082803.133243. PMID15952900.
- ^ Kastrinos F, Syngal S (2011). "Síndromes de cáncer colorrectal heredado". Cáncer J 17 (6): 405 – 15. Doi:10.1097/PPO.0b013e318237e408. PMC3240819. PMID22157284.
- ^ HP Neumann, Pawlu C, Peczkowska M et al distintas características clínicas del paraganglioma síndromes asociados con SDHB y SDHD mutaciones genéticas. JAMA. 2004; 292 (8): 943. PMID 15328326
- ^ Malkin d. (abril de 2011) "El síndrome de Li-fraumeni". Los genes cáncer 2 (4): 475 – 84. Doi:10.1177/1947601911413466. PMC3135649. PMID21779515.
- ^ Bakry, D (2013). P53 en la clínica: las mutaciones de línea germinal TP53: genética del síndrome de Li-Fraumeni. Nueva York: Springer. págs. 167-188. ISBN978-1-4614-3676-8.
- ^ Abedul JM (julio de 1994). Síndromes de cáncer familiar y racimos. Br. med Bull. 50 (3): 624 – 39. PMID7987644.
- ^ Quesnel S, Malkin D (agosto de 1997). "La predisposición genética al cáncer y síndromes de cáncer familiar". Pediatr. Clin. Soy Norte. 44 (4): 791-808. Doi:10.1016/s0031-3955 (05) 70530-7. PMID9286285.
- ^ Sampson JR, Jones N (2009). "Poliposis asociada a MUTYH". Best Pract Res Clin Gastroenterol 23 (2): 209 – 18. Doi:10.1016/j.BPG.2009.03.006. PMID19414147.
- ^ Manfredi M, Vescovi P, Bonanini M, Porter S (marzo de 2004). "Síndrome del carcinoma basocelular nevoide: una revisión de la literatura". Int J Oral Surg gérmenes 33 (2): 117 – 24. Doi:10.1054/ijom.2003.0435. PMID15050066.
- ^ Lo Muzio L (2008). "Síndrome (síndrome de Gorlin) del carcinoma basocelular nevoide". Orphanet J rara Dis 3:: 32. Doi:10.1186/1750-1172-3-32. PMC2607262. PMID19032739.
- ^ Richard, S; Gardie, B; Couvé, S; Gad, S (30 de mayo de 2012). "Enfermedad de von Hippel-Lindau: Cómo una enfermedad rara ilumina la biología del cáncer". Seminarios en biología del cáncer 23 (1): 26 – 37. Doi:10.1016/j.semcancer.2012.05.005. PMID22659535.
- ^ Henry, Todd; Campell, James; Hawley, Arthur (1969). Diagnóstico clínico de Todd-Sanford por métodos de laboratorio, editado por Israel Davidsohn [y] John Bernard Henry. (14ª ed.). Philadelphia: Saunders. p. 555. ISBN0-7216-2921-0.
- ^ Wong WT, E n, Agró Coleman HR et al (febrero de 2007). "Correlación genotipo – fenotipo en la enfermedad de von Hippel-Lindau con angiomatosis retiniana". Archivos de Oftalmología. 125 (2): 239-45. Doi:10.1001/archopht.125.2.239. PMC3019103. PMID17296901. de 2008-10-22.
- ^ AR Lehmann, D McGibbon, Stefanini M (2011). "Xerodermia pigmentosa". Orphanet J rara Dis 6:: 70. Doi:10.1186/1750-1172-6-70. PMC3221642. PMID22044607.
- ^ Niedernhofer LJ, Bohr VA, Sander M, Kraemer KH (2011). "Reparación de Xerodermia pigmentosa y otras enfermedades del envejecimiento humano y ADN: las moléculas a los pacientes". Mech. envejecimiento dev. 132 (6-7): 340 – 7. Doi:10.1016/j.Mad.2011.06.004. PMC3474983. PMID21708183.