Antagonista de los receptores CCR5
|
|
Este artículo puede requieren limpieza para satisfacer de Copro estándares de calidad. Es el problema específico: el artículo es demasiado detallado en un solo subtema (75% texto en moléculas pequeñas, descubrimiento de fármacos preclínica), basado en gran parte en primaria encendido, y también su selección y resumen o Por favor ayuda mejorar este artículo Si se puede. (De diciembre de 2014) |
|
|
Este artículo sólo describe un aspecto altamente especializado de su tema asociado. (De diciembre de 2014) |
|
|
Este artículo se basa demasiado en referencias Para fuentes primarias. (De diciembre de 2014) |
|
|
Este artículo posiblemente contiene investigación original. (De diciembre de 2014) |
Antagonistas del receptor CCR5 son una clase de pequeñas moléculas que antagonizar a el Receptor CCR5. El C-C motivo chemokine receptor CCR5 está involucrado en el proceso por el cual VIH, el virus que causa AYUDAS, entra en las células. Por lo tanto son los antagonistas de este receptor inhibidores de la entrada y tienen potenciales aplicaciones terapéuticas en el tratamiento de infecciones por el VIH.
El ciclo de vida del VIH presenta objetivos potenciales para la terapia de droga, uno de ellos es la vía de entrada viral. Los receptores de C-C motivo chemokine CCR5 y CXCR4 son los principales Chemokine receptores implicados en el proceso de entrada del VIH. Estos receptores pertenecen a la siete (transmembrana del receptor g-proteína-juntadoGPCR) familia y son predominantemente expresado en humanos Células T, células dendríticas y macrófagos, Células de Langerhans.[1] Juegan un papel importante como Co de los receptores que el VIH tipo 1 (VIH-1) utiliza para unir a las células antes de la fusión viral y entrada en las células del huésped.[1] Aislamientos de VIH pueden dividirse en R5 y X 4 cepas. R5 es cuando el virus usa el co-receptor CCR5 y filtre el X 4 es cuando utiliza CXCR4.[2] La ubicación de los receptores CCR5 en la superficie celular, moléculas grandes y pequeñas tienen el potencial de interferir con la interacción viral CCR5 y inhibir la entrada viral en las células humanas.[3]
Contenido
- 1 Historia
- 2 Mecanismo de acción
- 3 Desarrollo de fármacos
- 3.1 Aplaviroc
- 3.2 Vicriviroc
- 3.3 Maraviroc
- 4 Pharmacophore
- 5 Enlace
- 5.1 Aplaviroc
- 5.2 Maraviroc
- 5.3 Aplaviroc
- 6 Otros antagonistas de CCR5
- 7 Véase también
- 8 Referencias
- 9 Acoplamientos externos
Historia
Desde el descubrimiento del VIH en la década de 1980, ha realizado notables progresos en el desarrollo de la novela medicamentos antivirales.[2] El gatillo para el descubrimiento de la CCR5 antagonistas de los fue la observación de que un pequeño porcentaje de poblaciones de alto riesgo demostró cualquier resistencia o retraso en el desarrollo de la enfermedad. Esta población fue encontrada para tener un mutación de (CCR5-Δ32) en el gen que codifica para el receptor CCR5, que se traduce en resistencia casi completa frente al VIH-1 infección y científicos luego descubrieron el papel clave de la receptores de superficie celular CCR5 y CXCR4 exitosa fusión viral e infección.[4] En 1996, fue demostrado que CCR5 sirve como un co-receptor para la mayoría comúnmente transmite las cepas VIH-1, R5. Este tipo de virus es predominante durante las primeras etapas de la infección y sigue siendo el dominante forma en más del 50% de la última etapa de pacientes infectados por VIH-1,[5][6] sin embargo las cepas R5 pueden desarrollarse eventualmente en X 4 la progresión de la enfermedad.[2] Esta información llevó al desarrollo de una nueva clase de medicamentos contra el VIH llamados a antagonistas de CCR5.[7]
Mecanismo de acción

VIH entra en las células del huésped en la sangre adjuntándose a receptores de en la superficie de la Células CD4 +.[8] Entrada viral a la célula CD4 + se inicia con el accesorio de la R5 VIH-1 glucoproteína 120 (gp120) para el receptor del T-cell de CD4 +, que produce un cambio conformacional en gp120 y permite enlazar a CCR5, tal modo disparo (glicoproteína 41gp41) mediada por la fusión de la envoltura vírica con el membrana de la célula y de la nucleocápside entra en la célula huésped (Figura 1).[8][9] Antagonistas del correceptor CCR5 previenen el VIH-1 de entrar y de infectar células inmunes mediante el bloqueo del receptor de superficie celular CCR5.[10] Pequeño molécula de los antagonistas de CCR5 se unen a una hidrofóbico bolsillo formado por la transmembrana hélices del receptor CCR5.[11] Se piensa que interactúan con el receptor en un alostéricos forma de fijación al receptor en una conformación que prohíbe su función co-receptor.[12]
Desarrollo de fármacos
|
|
En esta sección necesita referencias adicionales para verificación. (De diciembre de 2014) |
Como se mencionó, el receptor CCR5 es un receptor proteína G acoplado (GPCR). Antes del descubrimiento del papel de CCR5 en la infección por VIH, muchas compañías farmacéuticas ya construyó una sustancial colección de compuestos blanco GPCRs.[citación necesitada] Algunos de estos compuestos sería punto de partida para antagonista de CCR5 química medicinal, pero necesita optimización para mejorar la selectividad de CCR5 y potencia y para mejorar la farmacocinética propiedades.[citación necesitada] Un problema importante fue la afinidad de disponible proyección hits para la hERG canal del ion;[citación necesitada] inhibición de hERG conduce a Intervalo QT prolongación, que puede aumentar el riesgo de desarrollar fatal arritmias ventriculares.[3][13] Muchos antagonistas de CCR5 han sido estudiados por las empresas farmacéuticas, pero pocos de ellos realmente han alcanzado estudios de eficacia humana; por ejemplo AstraZeneca,[14][fuente primaria no es necesitada] Novartis,[15][fuente primaria no es necesitada] Merck,[16][fuente primaria no es necesitada] y Takeda[17][fuente primaria no es necesitada] han utilizado su compuesto GPRC-targeting colecciones para desarrollar un potente antagonista de CCR5, pero ninguno de ellos han llegado a ensayos clínicos.[citación necesitada] Tres compañías farmacéuticas estaban en competencia para ser los primeros en tener una pequeña molécula antagonista CCR5 aprobado:[citación necesitada] GlaxoSmithKline(GSK) con su compuesto aplaviroc,[citación necesitada] Schering-Plough con Vicriviroc,[citación necesitada] y Pfizer con maraviroc.[citación necesitada] Todos los compuestos alcanzaron ensayos clínicos en seres humanos;[citación necesitada] sólo maraviroc ha sido aprobado[vago] por la Administración de drogas y alimentos de Estados Unidos (FDA).[3] En la siguiente sección se discutirá el desarrollo de estos tres compuestos.
Aplaviroc

Aplaviroc se originó de una clase de derivados de spirodiketopiperazine. La figura 2 muestra la estructura molecular de lo compuestos de plomo y el aplaviroc compuesto final. El compuesto de plomo mostró buena potencia en el bloqueo de CCR5 en una serie de cepas de VIH R5 y contra cepas resistentes a múltiples fármacos.[3] El problema con este compuesto no era su selectividad de CCR5, pero la biodisponibilidad oral.[3][18] Esto condujo al desarrollo de la molécula y el resultado fue un compuesto llamado aplaviroc. Lamentablemente a pesar de los prometedores resultados clínicos preclínicos y precoces algunos toxicidad hepática severa se observó en el tratamiento de la ingenua y pacientes que llevaron a la interrupción en el desarrollo de aplaviroc.[3]
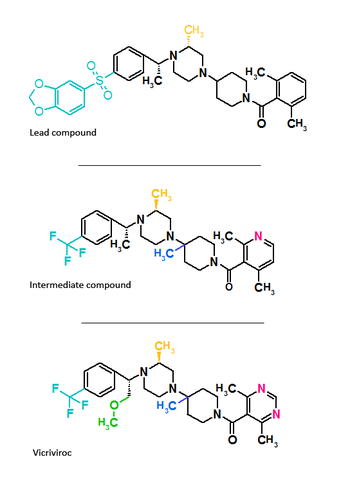
Vicriviroc
Schering-Plough identificada un compuesto activo durante la proyección.[3] La figura 3 muestra la estructura molecular del plomo compuesto, intermedio compuesto y el compuesto final Vicriviroc. El compuesto de plomo que contenía un piperazina Andamios y era un potente receptor muscarínico (M2) antagonista con actividad moderada de CCR5. Los cambios que se hicieron en el lado izquierdo del plomo del compuesto y la adición de un grupo metilo en el grupo de la piperazina ((S)-methylpiperazine) resultó en el compuesto intermedio que tuvo buena afinidad para los receptores CCR5, pero muy poca afinidad por muscarinic actividad, sin embargo, el compuesto mostró afinidad por el canal hERG del ion.[19][20] Reconstrucción posterior condujo al desarrollo de la final compuesto vicriviroc, cuando Schering descubrió que el piridil N-óxido en el intermedio podría ser reemplazado por 4, 6-dimetilpirimidina carboxamida. Vicriviroc tuvo una excelente selectividad para CCR5 receptores muscarínicos y hERG afinidad fue reducido grandemente.[21][22] Ensayo clínico de fase I de vicriviroc dio resultados prometedores, por lo que se inició un estudio fase II en el tratamiento de pacientes. El estudio fase II se suspendió ya que hubo un avance viral en el grupo de vicriviroc en comparación con el Grupo de control. Estos resultados sugirieron que vicriviroc no era eficaz en el tratamiento de pacientes de tratamiento en comparación con los tratamientos actuales. Se realizó otro estudio fase II clínica en pacientes. Los resultados fueron que vicriviroc tenía actividad antiviral fuerte pero cinco instancias de cáncer entre los participantes se informaron, sin embargo, el estudio fue continuado ya que la falta de asociación causal de la tumores malignos y vicriviroc.[3] A finales de 2009, vicriviroc fue reportado por la empresa que han entrado en estudios de fase II en el tratamiento de pacientes y estudios de fase III en pacientes.[23][mejor fuente es necesitada]
Maraviroc
Pfizer se dirigió a proyección de alto rendimiento en su búsqueda de un buen punto de partida para un antagonista de la pequeña molécula CCR5. Su investigación dio lugar a un compuesto que presenta afinidad débil y sin actividad antiviral, pero representa un buen punto de partida para la mayor optimización.[3] Compuestos 1-9 en la tabla 1 muestran el desarrollo de maraviroc en pocos pasos. La estructura química de la molécula de partida se presenta como compuesto 1. Su primer objetivo era minimizar CYP2D6 actividad de la molécula y a reducir su lipofilia. Sustituye el imidazopyridine con bencimidazol y el grupo de benzhydril fue intercambiado hacia fuera para un Benzamida. El resultado fue de 2 compuestos.[3] Este compuesto mostró potencia buen atascamiento y el inicio de una actividad antiviral. (SAR) másrelación estructura-actividad) optimización de la amida región e identificar los enantiomérica preferencia condujo a la estructura amida cyclobutyl compuesto 3. Sin embargo, el problema con la actividad de CYP2D6 del compuesto era aún inaceptable por lo que tuvieron que realizar más optimización de SAR que determinó que el [3.2.1]-azabicycloamine (topane) podría sustituir la parte de la aminopiperidine. Este cambio en la estructura química condujo a compuesto 4. Compuesto 4 no tenía ninguna actividad de CYP2D6 conservando excelente afinidad y antiviral actividad.[3][24] Aunque 4 compuesto mostró resultados prometedores demostrado inhibición del 99% en el hERG canal del ion. Que la inhibición es inaceptable ya que puede conducir a Intervalo QTc prolongación. El equipo de investigación hizo algunas modificaciones para ver que parte de la molécula desempeñó un papel en la afinidad de hERG. Compuesto 5 muestra un análogo que sintetiza que contenía una cabeza de puente de oxígeno en la tropano anillo; sin embargo, la reconstrucción no tuvo efecto en la afinidad de hERG.[25] Enfoca entonces en la superficie polar en la molécula para marcar la afinidad de hERG. Estos esfuerzos resultaron en compuesto 6. Ese compuesto conservada deseada actividad antiviral y selectiva contra la inhibición de hERG pero el problema era su biodisponibilidad. Reducción de la lipofilia, mediante la sustitución del grupo benzimidazol con un grupo de triazoles sustituidos dio compuesto 7. 7 compuesto había demostrado una reducción significativa en la lipofilia y mantuvo la actividad antiviral pero de nuevo, con la introducción de un grupo de cyclobutyl el compuesto demostrado inhibición de hERG. Cambiar el tamaño del anillo en 7 compuesto de una unidad cyclobutyl a una unidad cyclopentyl compuesto 8, llevado a un aumento significativo en la actividad antiviral y la pérdida de afinidad de hERG. Desarrollo posterior condujo al descubrimiento de un 4, 4'-difluorocyclohexylamide también conocido como maraviroc. Maraviroc había conservado excelente actividad antiviral, aunque no demostrando ninguna afinidad significativa hERG. La falta de afinidad de hERG fue predicha por el gran tamaño del grupo cyclohexyl y la alta polaridad de los sustitutos de fluoro.[3][24][25] En agosto de 2007 la FDA aprobó al primer antagonista de CCR5, maraviroc, descubierto y desarrollado por Pfizer.[4][7]
 |
 |
 |
| Compuesto 1 | Compuesto 2 | Compuesto 3 |
 |
 |
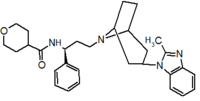 |
| Compuesto 4 | Compuesto de 5 | Compuesto 6 |
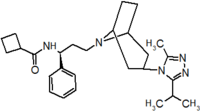 |
 |
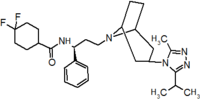 |
| Compuesto 7 | Compuesto de 8 | Compuesto de 9 (Maraviroc) |
Pharmacophore

El predictivo Pharmacophore modelo fue desarrollado para una gran serie de piperidina- y piperazina-basado en antagonistas de CCR5 por Schering-Plough Research Institute. Su hipótesis consistió en sobre todo cinco características, dos aceptores de hidrógeno, marcadas C y D en Figura 4 y tres hidrofóbico grupos A, B y E en la figura 4. Parte B generalmente tiene un grupo de nitrógeno básico. El modelo se validó utilizando diversos conjunto de seis antagonistas de CCR5 de cinco diferentes compañías farmacéuticas. El mejor modelo predijo correctamente estos compuestos como altamente activa. Es posible utilizar el modelo como una herramienta de proyección virtual de nuevos antagonistas de CCR5 moleculares pequeño y también para predecir la actividad biológica de compuestos antes de emprender su síntesis costosos.[26]
Enlace
CCR5 es miembro de g-proteína-juntados, siete transmembrana segmento de receptores. La estructura del receptor comprende siete-helix bundle en la región transmembrana, estas regiones se etiquetan VII en la figura 5 y 6. Los antagonistas de CCR5 se predicen enlazar a un bolsillo de unión supuesta que está enterrado dentro del dominio transmembrana, rodeado por las siete hélices transmembranales. El bolsillo de Unión es muy hidrofóbico con múltiples residuos aromáticos forro del bolsillo. Los residuos principales son triptófano 86 y 248 (Trp86, Trp248), tirosina 108 y 251 (Tyr108, Tyr251), fenilalanina 109 (phe109), treonina 195 (Thr195), isoleucina 198 (Ile198), ácido glutámico 283 (Glu283). Antagonistas de CCR5 son muy diferentes en forma y electrostático potencial, aunque todos ellos comparten el mismo bolsillo de Unión. Lo interesante de la Unión de estas moléculas es que exhiben modos de encuadernación significativamente diferentes, aunque todos ellos establecen una red de amplia interacción con CCR5.[27][28][29]
Aplaviroc
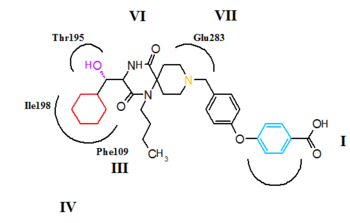

El modo de vínculo putativo de aplaviroc se muestra en la figura 5. La interacción clave saltbridge entre aplaviroc y Glu283 se predice para ser muy débil en comparación con otros antagonistas de CCR5. El grupo del oxhidrilo en aplaviroc forma un fuerte enlace de hidrógeno a los residuos polares Thr195. Esta interacción de enlace de H es el más fuerte con aplaviroc en comparación con otros antagonistas de CCR5. Cyclohexyl grupo en la estructura de aplaviroc se prevé interactuar con el receptor en un bolsillo hidrofóbico formado por Ile198, Thr15 y Phe109 y se cree que Mostrar interacciones hidrofóbicas bastante fuertes. Los investigadores predicen que el grupo t-butilo de aplaviroc se entierra dentro del paquete helicoidal a través de fuerte interacción hidrófoba con múltiples residuos aromáticos del receptor CCR5.[28]
Maraviroc
El modo putativo de maraviroc se muestra en la figura 6. La interacción más fuerte se estima entre maraviroc y ácido glutámico (Glu283) a través de una fuerte interacción de puente de sal. La interacción entre el triptófano (Trp86) y maraviroc involucra en forma de T π-π de apilamiento mientras que la interacción con fenilalanina (Phe109) se prevé que hydrophopic. Tirosina (Tyr108) está pensado para interactuar con el grupo fenil en maraviroc a través de una interacción desplazada paralelo. La interacción entre maraviroc e isoleucina (Ile198) se prevé que en su mayoría hidrofóbicas en la naturaleza y la interacción entre maraviroc y tirosina (Tyr251) es muy limitada.[28]
Aplaviroc
Aplaviroc tiene una característica única de la conservación de dos de los ligandos de la proteína chemokine natural a CCR5 y posterior activación, mientras que maraviroc y otros antagonistas de los bloquean casi totalmente las interacciones de la quimiocina CCR5. Este tipo de interferencia hasta ahora se considera seguro, e individuos que naturalmente carecen de CCR5 no muestran problemas obvios de salud. Sin embargo, para limitar la toxicidad y efectos secundarios de los antagonistas de CCR5 sería ideal para poder preservar la función del receptor de quimiocina. En consecuencia, debe ser de interés para el diseño de inhibidores que específicamente alteran CCR5-gp120 vinculante pero que no afectan a la activación del chemokine CCR5.[29]
Otros antagonistas de CCR5

Desarrollo de nuevos antagonistas de CCR5 continúa, tanto por sus efectos antivirales y también de utilidad potencial en una variedad de autoinmune indicaciones. Investigadores de Ono han descubierto una nueva serie de potentes antagonistas de pequeña molécula CCR5. Optimización de plomo fue perseguido por equilibrar las tendencias opuestas de estabilidad metabólica y de la potencia. Combinación de la plantilla de spiropiperidine con elementos de pharmacophore de aplaviroc y programa de antagonista de CCR5 de Schering, llevó a lo compuesto de plomo inicial en esta serie. Desarrollo posterior de ese compuesto de plomo condujo al descubrimiento del compuesto A en la figura 7 - un compuesto que posee una buena selectividad y propiedades farmacocinéticas.[30]
El antagonista de CCR5 INCB009471 ha nanomolares actividad contra el VIH-1 in vitro. Este compuesto demostró potente y prolongada actividad antiviral contra el VIH R5 trópico-1 cuando dada una vez al día dosis de 200 mg durante 14 días. Estos resultados apoyaron clínica desarrollo de INCB009471 y desde entonces han progresado a los ensayos clínicos de fase IIb. A partir de 2009 el estudio de este compuesto es inactivo y no estudios adicionales están proyectados en este momento.[31]
Véase también
- Cenicriviroc
- CD4
- CCL5
- CCR5
- Subtipos del VIH
- Tropismo del VIH
- Descubrimiento y desarrollo de los inhibidores no nucleósidos de la transcriptasa reversa
- Descubrimiento y desarrollo de inhibidores de la transcriptasa inversa análogos de los nucleósidos y nucleótidos
Referencias
- ^ a b Lederman mm., Penn-Nicholson, M Cho, Mosier D (agosto de 2006). "Biología de CCR5 y su papel en la infección por el VIH y el tratamiento". JAMA 296 (7): 815-26. doi:10.1001/Jama.296.7.815. PMID16905787.
- ^ a b c De Clercq E (diciembre de 2007). "El diseño de fármacos para el VIH y VHC". Naturaleza comentarios Drug Discovery 6 (12): 1001-18. doi:10.1038/nrd2424. PMID18049474.
- ^ a b c d e f g h i j k l Polea, Shon (2007). "antagonistas de CCR5: desde el descubrimiento de la eficacia clínica". En Neote, Kuldeep; Letts, Gordon L.; Moser, Bernhard. Chemokine biología, Investigación básica y aplicación clínica 2. Birkhäuser Basilea. págs. 145 – 163. doi:10.1007/978-3-7643-7437-2_11. ISBN978-3-7643-7195-1.
- ^ a b Lalezari, J.; Goodrich, J.; DeJesus, E.; Lampiris, H.; Gulick, R.; Saag, M.; Ridgway, C.; McHale, M.; Van Der Ryst, E. "eficacia y seguridad de Maraviroc más optimizado fondo terapia en Viremic, ART-Experienced pacientes infectados con CCR5-trópico HIV‑1, resultados de 24 semanas de un estudio de fase 2b/3 en los Estados Unidos y el Canadá. 104bLB Resumen". 14 conferencia sobre retrovirus e infecciones oportunistas.
- ^ Samson M, Libert F, Doranz BJ; et al (agosto de 1996). "Resistencia a la infección por VIH-1 en individuos caucásicos con alelos del mutante del gene del receptor de chemokine de CCR-5". Naturaleza 382 (6593): 722 – 5. doi:10.1038/382722a0. PMID8751444.
- ^ Dragic T, V Litwin, Allaway GP; et al (junio de 1996). "HIV-1 entrada en células CD4 + es mediada por el receptor de quimiocina CC-CKR-5". Naturaleza 381 (6584): 667 – 73. doi:10.1038/381667a0. PMID8649512.
- ^ a b C Flexner (diciembre de 2007). "desarrollo de medicamentos de VIH: los próximos 25 años". Naturaleza comentarios Drug Discovery 6 (12): 959 – 66. doi:10.1038/nrd2336. PMID17932493.
- ^ a b Ray N, Doms RW (2006). correceptores del VIH-1 y sus inhibidores. Asuntos actuales en Microbiología e Inmunología. Asuntos actuales en Microbiología e Inmunología 303:: 97-120. doi:10.1007/978-3-540-33397-5_5. ISBN978-3-540-29207-4. PMID16570858.
- ^ Westby M, van der Ryst E (2005). "antagonistas de CCR5: orientada a host antivirales para el tratamiento de la infección por VIH". Quimioterapia y química antiviral 16 (6): 339 – 54. PMID16329283.
- ^ Briz V, Poveda E, Soriano V (abril de 2006). "inhibidores de entrada del VIH: mecanismos de acción y resistencia de las vías". El diario de la quimioterapia antimicrobiana 57 (4): 619 – 27. doi:10.1093/JAC/dkl027. PMID16464888.
- ^ JD de Murga, Franti M, DC Pevear, Maddon PJ, Olson WC (octubre de 2006). "Potente antivirus sinergia entre anticuerpos monoclonales y los inhibidores de la pequeña molécula CCR5 del Virus de inmunodeficiencia humana tipo 1". Agentes antimicrobianos y quimioterapia 50 (10): 3289 – 96. doi:10.1128/AAC.00699-06. PMC1610098. PMID17005807.
- ^ Watson C, S Jenkinson, Kazmierski W, Kenakin T (abril de 2005). "el CCR5 receptor basado en mecanismo de acción de 873140, un potente alostéricos VIH inhibidor". Farmacología Molecular 67 (4): 1268-82. doi:10.1124/mol.104.008565. PMID15644495.
- ^ B Fermini, fosa AA (junio de 2003). "El impacto de la prolongación de QT medicamentosa intervalo de descubrimiento de fármacos y desarrollo". Naturaleza comentarios Drug Discovery 2 (6): 439 – 47. doi:10.1038/nrd1108. PMID12776219.
- ^ Cumming JG, Cooper AE, mugre K; et al (noviembre de 2005). "moduladores del receptor CCR5 humano. Parte 2: SAR de sustituir 1 phenylacetamides-(3,3-diphenylpropyl)-piperidinil ". Letras de Química bioorgánica y medicinales 15 (22): 5012 – 5. doi:10.1016/j.BMCL.2005.08.014. PMID16154744.[fuente primaria no es necesitada]
- ^ Thoma G, Nuninger F, M Schaefer, Akyel KG, Albert R, Beerli C, C Bruns, Francotte E, Luyten M, MacKenzie D, Oberer L, Streiff MB, Wagner T, Walter H, G Weckbecker, Zerwes HG. (Abril de 2004). "Oral bioavailable competitivo CCR5 antagonistas". Diario de la Química Medicinal 47 (8): 1939-55. doi:10.1021/jm031046g. PMID15055994.[fuente primaria no es necesitada]
- ^ Bryan Oates, Richard J. Budhu, lijadora G. molinos, Malcolm MacCoss, Lorena Malkowitz, Martin S. Springer, Bruce L. Daugherty, Sandra L. Gould, Julie a. DeMartino, Salvatore J. Siciliano, Anthony Carella, Gwen Carver, Karen Holmes, Renee Danzeisen, Daria Hazuda, Joseph Kessler, Janet Lineberger, Michael Miller, William A. Schleif, Emilio A. Emini (enero de 2001). "antagonistas del receptor CCR5 humano como agentes anti-VIH-1. Parte 1: descubrimiento y las relaciones estructura-actividad inicial de 1 - amino-2-phenyl-4-(piperidin-1-yl)butanes ". Letras de Química bioorgánica y medicinales 11 (2): 259-64. doi:10.1016/S0960-894 X (00) 00637-5. PMID11206473.[fuente primaria no es necesitada]
- ^ Tremblay CL Giguel F, Guan Y, Chou TC, Takashima K, Hirsch MS (agosto de 2005). "TAK-220, un antagonista del CCR5 de molécula pequeña novela, tiene interacciones de Virus de inmunodeficiencia humana Favorable con otros antirretrovirales In Vitro". Agentes antimicrobianos y quimioterapia 49 (8): 3483 – 5. doi:10.1128/AAC.49.8.3483-3485.2005. PMC1196290. PMID16048964.[fuente primaria no es necesitada]
- ^ Maeda K, H de Nakata, Koh Y; et al (agosto de 2004). "Inhibidor de CCR5 basada en Spirodiketopiperazine que conserva las interacciones CC-Chemokine/CCR5 y ejerce actividad potente contra R5 Virus de inmunodeficiencia humana tipo 1 In Vitro". Diario de la virología 78 (16): 8654 – 62. doi:10.1128/JVI.78.16.8654-8662.2004. PMC479103. PMID15280474.
- ^ Tagat JR, McCombie SW, Steensma RW; et al (agosto de 2001). "antagonistas de CCR5 base de piperazina como inhibidores de VIH-1. I: 2 (S)-piperazina de metilo como un elemento clave de pharmacophore ". Letras de Química bioorgánica y medicinales 11 (16): 2143-6. doi:10.1016/S0960-894 00381-X (01). PMID11514156.
- ^ Tagat JR, RW Steensma, McCombie SW; et al (octubre de 2001). "Antagonistas de CCR5 base de piperazina como inhibidores de VIH-1. II. descubrimiento de carbonilo 1-[(2,4-dimethyl-3-pyridinyl)] -4-piperidina methyl-4-[3(S)-methyl-4-[1(S)-[4-(trifluoromethyl)phenyl]ethyl]-1-piperazinyl]-N1-óxido (Sch-350634), un oral biodisponible, potente antagonista CCR5 ". Diario de la Química Medicinal 44 (21): 3343-6. doi:10.1021/jm0155401. PMID11585438.
- ^ McCombie SW, Tagat JR, Vicepresidente de SF; et al (febrero de 2003). "antagonistas de CCR5 base de piperazina como inhibidores de VIH-1. III: síntesis, antivirales y farmacocinéticos perfiles de carboxamides heteroaryl simétrico ". Letras de Química bioorgánica y medicinales 13 (3): 567-71. doi:10.1016/S0960-894 X (02) 00918-6. PMID12565973.
- ^ Tagat JR, McCombie SW, Nazareno D; et al (mayo de 2004). "antagonistas de CCR5 base de piperazina como inhibidores de VIH-1. IV. descubrimiento de carbonilo 1-[(4,6-dimethyl-5-pyrimidinyl)]-4-[4-[2-methoxy-1(R)-4-(trifluoromethyl)phenyl]ethyl-3(S)-methyl-1-piperazinyl]-4-methylpiperidine (Sch-417690/Sch-D), un potente y altamente selectivo y oral bioavailable CCR5 antagonista ". Diario de la Química Medicinal 47 (10): 2405 – 8. doi:10.1021/jm0304515. PMID15115380.
- ^ "Schering-Plough informes datos de Vicriviroc a largo plazo de extensión de etiqueta abierta fase II estudio en infectados por el VIH pacientes" (Comunicado de prensa). Schering-Plough. 14 de septiembre de 2009. 8 de noviembre 2009.[mejor fuente es necesitada]
- ^ a b A madera, armadura D (2005). "el descubrimiento de la antagonista de los receptores CCR5, UK-427.857, un nuevo agente para el tratamiento de la infección por el VIH y el SIDA". Avances en Química Medicinal. Avances en Química Medicinal 43: 239 – 71. doi:10.1016/S0079-6468 (05) 43007-6. ISBN978-0-444-51572-8. PMID15850827.
- ^ a b Precio DA, D de armadura, de Groot M; et al (septiembre de 2006). "Superar la afinidad HERG en el descubrimiento del maraviroc de antagonista de CCR5". Letras de Química bioorgánica y medicinales 16 (17): 4633-7. doi:10.1016/j.BMCL.2006.06.012. PMID16782336.
- ^ Debnath AK (octubre de 2003). "generación de pharmacophore predictivo los modelos para antagonistas de CCR5: estudio con compuestos a base de piperazina y piperidina como una nueva clase de inhibidores de la entrada HIV-1". Diario de la Química Medicinal 46 (21): 4501 – 15. doi:10.1021/jm030265z. PMID14521412.
- ^ Maeda K, Das D, Ogata-Aoki H; et al (mayo de 2006). "estructurales y moleculares las interacciones de los inhibidores de CCR5 con CCR5". El diario de química biológica 281 (18): 12688-98. doi:10.1074/jbc. M512688200. PMID16476734.
- ^ a b c R Kondru, Zhang J, Ji C; et al (marzo de 2008). "Interacciones moleculares de CCR5 con clases principales de los antagonistas de CCR5 de pequeñas moléculas anti-VIH". Farmacología Molecular 73 (3): 789-800. doi:10.1124/mol.107.042101. PMID18096812.
- ^ a b Wang T, Duan Y (junio de 2008). "atar los modos de entrada inhibidores de CCR5 dirigiéndose VIH: antagonistas parciales y totales". Diario de gráficos moleculares y modelos 26 (8): 1287-95. doi:10.1016/j.jmgm.2007.12.003. PMC2701198. PMID18249144.
- ^ Rotstein DM, Gabriel SD, Makra F; et al (septiembre de 2009). "Antagonistas de Spiropiperidine CCR5". Letras de Química bioorgánica y medicinales 19 (18): 5401 – 6. doi:10.1016/j.BMCL.2009.07.122. PMID19674898.
- ^ Kuritzkes DR (marzo de 2009). "HIV-1 entrada Inhbitors: una visión general". Opinión actual en VIH y SIDA 4 (2): 82-7. doi:10.1097/COH.0b013e328322402e. PMC2753507. PMID19339945.
Acoplamientos externos
- Vídeo y texto de un documental de PBS sobre el descubrimiento de CCR5
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Los receptores beta-chemokine CCR3 y CCR5 facilitan la infección por cepas de VIH-1 primarios.
H Choe, de Farzan M, de sol Y, de Sullivan N, de B Rollins, de Ponath PD, de Wu L, de Mackay CR, G LaRosa, Newman W, N de Gerard, Gerard C, Sodroski J.
Célula. 1996 28 de Jun; 7:1135-48.