Reacción en cadena de polimerasa
|
|
Se ha sugerido que Aplicaciones de la PCR ser combinado en este artículo. (Discutir) Propuestos desde junio de 2013. |

El reacción en cadena de polimerasa (POLIMERIZACIÓN EN CADENA) es una tecnología en biología molecular utilizado para ampliar una copia única o unas pocas copias de un pedazo de DNA a través de varios órdenes de magnitud, generando miles de millones de copias de un determinado Secuencia de la DNA.
Desarrollado en 1983 por Kary Mullis,[1][2] PCR es ahora una técnica común y a menudo indispensable utilizada en laboratorios de investigación médicos y biológicos para una variedad de aplicaciones.[3][4] Estos incluyen Clonación de ADN para secuencia, Basado en el ADN filogenia, o análisis funcional de genes; el diagnóstico de enfermedades hereditarias; la identificación de huellas digitales genéticas (se utiliza en ciencias forenses y pruebas de paternidad); y la detección y el diagnóstico de enfermedades infecciosas. En 1993 Mullis fue concedido el Premio Nobel de química a lo largo de con Michael Smith por su trabajo en PCR.[5]
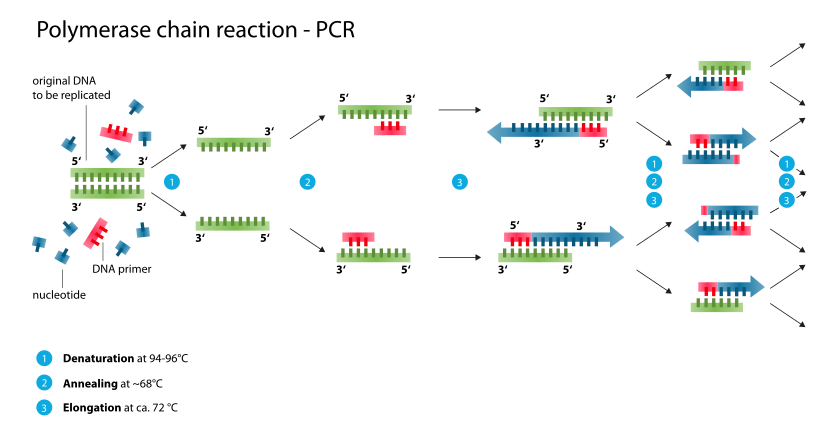
El método se basa en ciclos térmicos, que consiste en ciclos de calefacción y enfriamiento de la reacción repetida ADN de fusión y enzimática replicación de la DNA. Cartillas de (corta fragmentos de ADN) que contienen secuencias complementarias a la región de destino a lo largo con un Polimerasa de la DNA, que el método se nombra después, son componentes clave para permitir la amplificación selectiva y repetida. Como progresa PCR, el ADN generado es utilizado como plantilla para la replicación, pone en marcha un reacción en cadena en el que la plantilla de DNA es manera exponencial amplificado. PCR se puede modificar extensivamente para realizar una amplia gama de manipulaciones genéticas.
Casi todas las aplicaciones de PCR emplean una polimerasa DNA termoestable, tales como Taq polimerasa (una enzima aislada originalmente de la bacteria Aquaticus de Thermus). Esta polimerasa de la DNA enzimáticamente monta un nuevo filamento de ADN de bloques de construcción de ADN, la nucleótidos, usando el ADN como una plantilla y el ADN oligonucleótidos (también llamado Cebadores de ADN), que son necesarios para la iniciación de la síntesis de ADN. Uso de la mayoría de los métodos de PCR ciclos térmicos, es decir, alternativamente calentamiento y enfriamiento de la muestra PCR a través de una serie definida de pasos de temperatura.
En el primer paso, las dos hebras de la doble hélice de ADN se separan físicamente a una alta temperatura en un proceso llamado ADN de fusión. En el segundo paso, la temperatura se baja y se convierten en las dos hebras de ADN plantillas para polimerasa de la DNA amplificar selectivamente el ADN diana. Resultados de la selectividad de la polimerización en cadena de la utilización de cartillas de son complementarias a la región de ADN para amplificación en las condiciones específicas de ciclismo térmicas.

Contenido
- 1 Principios y procedimiento
- 1.1 Procedimiento
- 1.2 Etapas
- 2 Optimización
- 3 Aplicaciones
- 3.1 Aislamiento selectivo de ADN
- 3.2 Amplificación y cuantificación de ADN
- 3.3 Diagnóstico de enfermedades
- 3.4 Limitaciones
- 4 Variaciones
- 5 Historia
- 5.1 Controversias sobre patentes
- 6 Referencias
- 7 Enlaces externos
Principios y procedimiento


PCR amplifica una región específica de un filamento de la DNA (el destino del ADN). Métodos PCR más típicamente amplifican fragmentos de ADN de entre 0,1 y 10 kilo pares de bases (kbp), aunque algunas técnicas permiten la amplificación de fragmentos de hasta 40 kbp en tamaño.[6] La cantidad de producto amplificado se determina por los sustratos disponibles en la reacción, que se convierten en limitantes conforme avanza la reacción.[7]
Una PCR básica establecer requiere varios componentes y reactivos.[8] Estos componentes incluyen:
- Plantilla de la DNA contiene la región de la DNA (destino) para amplificar
- Dos cartillas de son complementarias a la 3' extremos (tres primeros) de cada uno de los sentido y anti-sentido filamento de la blanco de la DNA
- Taq polimerasa u otra Polimerasa de la DNA con una temperatura óptima alrededor de 70 ° c
- Deoxynucleoside trifosfatos (dNTPs, a veces llamado "Deoxinucleótidos trifosfatos"; nucleótidos contiene grupos trifosfato), los bloques de construcción de la cual la ADN polimerasa sintetiza una nueva hebra de DNA
- Solución tampón, proporciona un ambiente químico adecuado para optimizar la actividad y estabilidad de la ADN polimerasa
- Bivalente cationes, magnesio o manganeso iones; generalmente Mg2+ se utiliza, pero Mn2+ puede ser utilizado para DNA mediada por PCR mutagénesis, como mayor Mn2+ concentración aumenta la tasa de error durante la síntesis de ADN[9]
- Catión monovalente potasio iones
La PCR se realiza comúnmente en un volumen de reacción de 10-200 µL en tubos de reacción pequeña (0,2 – 0,5 volúmenes de ml) en un termociclador. El termociclador calienta y enfría los tubos de reacción para alcanzar las temperaturas requeridas en cada paso de la reacción (véase abajo). Muchos ciclistas termales modernos hacen uso de la Efecto Peltier, que permite tubos de calefacción y refrigeración del bloque con la PCR simplemente invirtiendo la corriente eléctrica. Tubos de paredes delgadas de la reacción permiten conductividad térmica favorable para permitir un rápido equilibrio térmico. Más termales ciclistas han calentado las tapas para evitar la condensación en la parte superior del tubo de reacción. Termocicladores más falta una tapa de calefacción requieren una capa de aceite sobre la mezcla de reacción o una bola de cera dentro del tubo.
Procedimiento
Típicamente, PCR consiste en una serie de cambios de temperatura repetidos de 20 – 40, llamado ciclos, cada ciclo comúnmente consisten en 2 – 3 pasos discretos de la temperatura generalmente tres (figura siguiente). El ciclismo es a menudo precedido por un paso de la sola temperatura a alta temperatura (> 90 ° C) y seguido por un asimiento en el extremo para extensión de producto final o el breve almacenaje. Las temperaturas utilizadas y la cantidad de tiempo que se aplican en cada ciclo dependen de una variedad de parámetros. Éstos incluyen la enzima usada para la síntesis de ADN, la concentración de iones divalentes y dNTPs en la reacción y la fusión (temperaturaTm) de los cebadores.[10]
- Paso de inicialización(Requerido solamente para las polimerasas de la DNA que requieren la activación por calor caliente-empiece PCR.[11]): Este paso consiste en calentar la reacción a una temperatura de 94-96 ° C (o 98 ° C si se utilizan polimerasas extremadamente termoestables), que se lleva a cabo de 1 a 9 minutos.
- Paso de desnaturalización:: Este paso es el primer evento de ciclismo regular y consiste en calentar la reacción a 94-98 ° C durante 20-30 segundos. Que causa ADN de fusión de la plantilla de la DNA por interrumpir los enlaces de hidrógeno entre bases complementarias, produciendo moléculas de ADN monocatenario.
- Paso de recocido:: La temperatura de reacción se baja a 50 – 65 ° C durante 20-40 segundos permitiendo el recocido de los cebadores a la plantilla de ADN monocatenario. Esta temperatura debe ser lo suficientemente baja como para permitir hibridación de la cartilla a la hebra, pero lo suficientemente alta para la hibridación ser específicos, es decir, la cartilla debe enlazarse sólo a una parte de la plantilla perfectamente complementaria. Si la temperatura es demasiado baja, podría enlazar imperfectamente la cartilla. Si es demasiado alto, no pudo enlazar la cartilla. Normalmente la temperatura de recocido es aproximadamente 3 – 5 ° C por debajo de la Tm de los primers utilizados. Estable DNA – DNA hidrógeno se forman solamente cuando la secuencia cartilla coincide muy de cerca con la secuencia de la plantilla. La polimerasa se une al híbrido primer plantilla y comienza la formación de ADN.
- Paso de extensión/elongación:: La temperatura en este paso depende de la ADN polimerasa usada; Taq polimerasa tiene su óptimo actividad temperatura de 75-80 ° C,[12][13] y comúnmente se utiliza una temperatura de 72 ° C con esta enzima. En este paso la ADN polimerasa sintetiza una nueva hebra de DNA complementaria a la hebra molde de DNA mediante la adición de dNTPs complementarios a la plantilla en 5' a 3' dirección, condensando el 5'-grupo fosfato de los dNTPs con el 3'-grupo hidroxilo al final de la cadena naciente de ADN (extensible). El tiempo de extensión depende tanto de la ADN polimerasa que utiliza como en la longitud del fragmento de ADN a amplificar. Como una regla del pulgar, en su temperatura óptima, la polimerasa de la DNA se polimeriza 1 mil bases por minuto. Bajo condiciones óptimas, es decir, si no hay limitaciones debido a la limitación de los sustratos o reactivos, en cada paso de la extensión, la cantidad de blanco de ADN se duplica, llevando a la amplificación (geométrica) exponencial del fragmento específico de ADN.
- Elongación final:: Este paso solo de vez en cuando se realiza a una temperatura de 70 – 74 ° C (esta es la temperatura necesaria para la actividad óptima de polimerasas más utilizadas en PCR) 5-15 minutos después de la última PCR ciclo para asegurarse de que cualquier ADN restante está completamente extendida.
- Espera final:: Este paso 4 – 15 ° c por un tiempo indefinido puede ser empleado para el almacenamiento a corto plazo de la reacción.
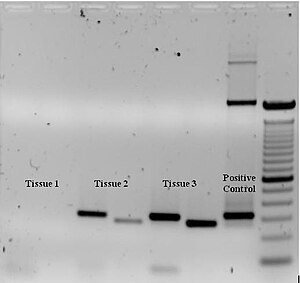
Para comprobar si la PCR generado el fragmento de ADN previsto (también a veces se denomina el amplimer o amplicones), electroforesis en gel de agarosa se emplea para la separación del tamaño de los productos PCR. Los tamaños de los productos PCR se determinaron por comparación con un Escalera de ADN (un marcador de peso molecular), que contiene fragmentos de ADN de tamaño conocido, en el gel junto con los productos PCR (ver Fig. 3).
Etapas
El proceso de polimerización en cadena puede dividirse en tres etapas:
Amplificación exponencial:: En cada ciclo se duplica la cantidad de producto (suponiendo que el 100% de eficiencia de la reacción). La reacción es muy sensible: sólo cantidades diminutas de ADN deben estar presentes.[14]
Nivelando la etapa:: La reacción se ralentiza como polimerasa de la DNA pierde actividad y consumo de reactivos como primers dNTPs provoca que se conviertan en limitantes.
Meseta de:: No más producto se acumula debido al agotamiento de los reactivos y enzimas.
Optimización
En la práctica, PCR puede fallar por varias razones, en parte debido a su sensibilidad a la contaminación que causa la amplificación de productos falsos de la DNA. Debido a esto, se han desarrollado una serie de técnicas y procedimientos para la optimización de las condiciones PCR.[15][16] Contaminación con ADN extraño se trata con protocolos de laboratorio y procedimientos que separan mezclas pre-PCR de los contaminantes potenciales del ADN.[8] Esto normalmente implica separación espacial de las áreas de PCR-instalación de áreas para el análisis o la purificación de los productos PCR, el uso de artículos desechables de plástico y limpiando cuidadosamente la superficie de trabajo entre configuraciones de reacción. Técnicas de diseño de la cartilla son importantes en la mejora de rendimiento del producto PCR y en evitar la formación de productos falsos y el uso de buffer alternativo componentes o enzimas de la polimerasa pueden ayudar con la amplificación de regiones problemáticas largo o de otro tipo de ADN. Adición de reactivos, tales como formamida, en buffer sistemas pueden aumentar la especificidad y el rendimiento de la PCR.[17] Simulaciones de computadora de teóricos (resultados PCRPCR electrónica) se puede realizar para ayudar en el diseño de la cartilla.[18]
Aplicaciones
Aislamiento selectivo de ADN
PCR permite aislamiento de fragmentos de la DNA de la DNA genomic por la amplificación selectiva de una región específica de ADN. Este uso de la PCR aumenta muchos métodos, tales como generación de sondas de hibridación para Sur o Norte hibridación y Clonación de ADN, que requieren grandes cantidades de ADN, que representa a una región específica de ADN. Polimerización en cadena suministros de estas técnicas con altas cantidades de ADN puro, lo que permite el análisis de muestras de ADN incluso de muy pequeñas cantidades de material de partida.
Otras aplicaciones de la PCR son Secuencia de la DNA determinar secuencias amplificadas por PCR desconocidas en que uno de la amplificación pueden usarse imprimadores en Sanger secuenciación, aislamiento de una secuencia de ADN para acelerar tecnologías de ADN recombinante, implicando la inserción de una secuencia de ADN en un plásmido, fago, o cósmido (dependiendo del tamaño) o el material genético de otro organismo. Colonias bacterianas (tales como E. coli) pueden ser examinados rápidamente por PCR para la DNA correcta Vector de construcciones.[19] PCR también puede ser usada para huella dactilar genética; una técnica forense para identificar a una persona o un organismo comparando DNAs experimentales a través de diferentes métodos basados en PCR.
Algunos métodos de 'huellas dactilares' de PCR tienen alto poder discriminativo y pueden utilizarse para identificar relaciones genéticas entre los individuos, como entre padres e hijos o entre hermanos y se utilizan en pruebas de paternidad (Fig. 4). Esta técnica puede utilizarse también para determinar relaciones evolutivas entre organismos cuando se utilizan ciertos relojes moleculares (es decir, la 16S rRNA y genes recA de microorganismos).[citación necesitada]

Amplificación y cuantificación de ADN
Porque PCR amplifica las regiones de ADN que se dirige, la PCR puede utilizarse para analizar cantidades extremadamente pequeñas de muestra. A menudo esto es crítico para Análisis forense, cuando sólo una cantidad de rastro de DNA está disponible como evidencia. PCR también puede utilizarse en el análisis de ADN antiguo es decenas de miles de años. Estas técnicas basadas en PCR han utilizado con éxito en animales, tales como 40 mil años mamuty también en el ADN humano, en aplicaciones que van desde el análisis del egipcio momias a la identificación de un Ruso Zar y el cuerpo del rey inglés Richard III.[20]
Métodos cuantitativos de PCR permiten la estimación de la cantidad de una secuencia dada presente en una muestra, una técnica que se aplica a menudo para determinar cuantitativamente los niveles de expresión del gen. PCR cuantitativa es una herramienta establecida para la cuantificación de ADN que mide la acumulación de productos de DNA después de cada ronda de amplificación por PCR.
Diagnóstico de enfermedades
PCR permite un diagnóstico precoz de malo enfermedades tales como leucemia y linfomas, que es actualmente el más alto-desarrollado en la investigación del cáncer y ya está siendo utilizado habitualmente. Ensayos de PCR pueden realizarse directamente en las muestras de ADN genómicas para detectar células malignas específicas de desplazamiento en una sensibilidad que es por lo menos 10.000 veces mayor que la de otros métodos.[citación necesitada]
PCR permite un diagnóstico rápido y altamente específico de enfermedades infecciosas, incluyendo ésos causados por bacterias o virus.[21] PCR también permite la identificación de microorganismos no cultivable o de crecimiento lento tales como micobacterias, bacterias anaerobias, o virus De cultivo de tejidos ensayos y modelos animales. La base para aplicaciones de diagnóstico de PCR en Microbiología es la detección de agentes infecciosos y la discriminación de no patógena de cepas patógenas en virtud de determinados genes.[21][22]
ADN viral puede detectarse además por PCR. Los cebadores utilizados deben ser específicos a las secuencias específicas en el ADN de un virus, y la PCR puede utilizarse para el análisis diagnósticos o secuencia de la DNA del genoma viral. La alta sensibilidad de PCR permite la detección de virus poco después de la infección y aún antes de la aparición de la enfermedad.[21] Tal detección temprana puede dar a los médicos un plazo significativo en tratamiento. La cantidad de virus ("carga viral") en un paciente también puede ser cuantificada mediante técnicas de cuantificación de DNA Polimerización en cadena-basado (véase abajo).
Limitaciones
Polimerasa de la DNA es propensa a errores, que a su vez provoca mutaciones en los fragmentos PCR que se realizan. Además, la especificidad de los fragmentos PCR puede mutar a la plantilla de ADN, debido a la Unión inespecífica de cartillas. Además es necesario para generar los iniciadores información previa sobre la secuencia.[23]
Variaciones
- PCR específica de alelo:: técnica de clonación o de un diagnóstico basado en variaciones del solo-nucleótido (SNVs no debe ser confundido con SNPs) (diferencias de base solo en un paciente). Requiere conocimiento previo de una secuencia de ADN, incluyendo las diferencias entre alelosy utiliza cebadores cuya 3' extremos abarcan el SNV (pares tampón alrededor de SNV generalmente incorporado). Amplificación por PCR bajo condiciones estrictas es mucho menos eficiente en presencia de un desajuste entre plantilla y cartilla, tan exitosa amplificación con una cartilla de SNP específico señales de presencia del SNP específico en una secuencia.[24] Ver Genotipado SNP para obtener más información.
- Asamblea PCR o Conjunto ciclismo polimerasa (PCA):: síntesis artificial de ADN larga secuencias mediante la realización de PCR en una piscina de oligonucleótidos largos con segmentos traslapados cortos. Los oligonucleótidos se alternan entre el sentido y antisentidas direcciones, y los segmentos traslapados determinan el orden de los fragmentos PCR, produciendo de tal modo selectivamente el producto largo final de ADN.[25]
- PCR asimétrico:: amplifica preferencial un filamento de la DNA de una plantilla de ADN bicatenario. Se utiliza en secuencia e hibridación donde se requiere la amplificación de solo uno de los dos filamentos complementarios. PCR se realiza como de costumbre, pero con un gran exceso de la cartilla para el filamento destinado a la amplificación. Debido a la lenta (aritmética) amplificación en la reacción después de que el primer limitante se ha utilizado para arriba, se requieren ciclos adicionales de PCR.[26] Una modificación reciente en este proceso, conocido como Linear-Aespués de-TJEOExponential-PCR (LATE-PCR), aplicaciones una cartilla limitadora con un más alto (temperatura fusiónTm) que el primer exceso para mantener la eficiencia de la reacción como la concentración de cartilla limitadora disminuye reacción mediados.[27]
- Polimerización en cadena dial-out:: un método altamente paralelo para recuperar precisas moléculas de ADN para la síntesis de genes. Una biblioteca compleja de moléculas de ADN se modifica con el únicos acompañamiento tags antes de secuenciación masivamente paralela. Cartillas de etiqueta dirigida entonces permiten la obtención de moléculas con secuencias deseadas por PCR.[28]
- PCR digital (dPCR):: utilizado para medir la cantidad de una secuencia de ADN en una muestra de ADN diana. La muestra de ADN es altamente diluida para que después de ejecutar muchos PCRs en paralelo, algunos de ellos no reciben una sola molécula de la ADN diana. El objetivo de concentración de ADN se calcula utilizando la proporción de resultados negativos. De ahí el nombre 'PCR digital'.
- Amplificación helicase-dependiente:: similar a la tradicional PCR, pero utiliza una temperatura constante en lugar de a través de ciclos de desnaturalización y recocido y extensión. DNA helicase, una enzima que desenrolla la DNA, se utiliza en lugar de la desnaturalización térmica.[29]
- Hot start PCR:: una técnica que reduce la amplificación inespecífica durante la inicial establecer las etapas de la polimerización en cadena. Se puede realizar manualmente calentando los componentes de la reacción a la temperatura de desnaturalización (p. ej., 95 ° C) antes de agregar la polimerasa.[30] Sistemas enzimáticos especializados han sido desarrollados que inhiben la actividad de la polimerasa en la temperatura ambiente, cualquiera por el atascamiento de un anticuerpo[11][31] o por la presencia de los inhibidores covalente que disocian solamente después de un paso de alta temperatura de activación. PCR Hot-start/frío-acabado se logra con nuevas polimerasas híbridas que son inactivas en la temperatura ambiente y se activan al instante a temperatura de elongación.
- En silico PCR (digital PCR, PCR, PCR electrónica, virtual e-PCR) se refiere a herramientas computacionales permite calcular resultados de reacción en cadena de polimerasa teórico utilizando un conjunto dado de cartillas de (puntas de prueba) para amplificar DNA secuencias de una secuencia genoma o transcriptoma. En silico PCR fue propuesto como una herramienta educativa para la biología molecular.[32]
- PCR intersequence-específico (ISSR): un método PCR para ADN fingerprinting que amplifica regiones entre simple secuencia se repite para producir una huella digital única de longitud de fragmentos amplificados.[33]
- Polimerización en cadena inversa:: comúnmente se utiliza para identificar las secuencias que flanquean alrededor genomic inserta. Se trata de una serie de Digestiones de DNA y ligadura del uno mismo, dando por resultado secuencias sabidas en cualquier final de la secuencia desconocida.[34]
- PCR ligadura-mediado:: usa pequeños linkers de ADN ligadas al ADN de interés y múltiples cebadores recocido a los linkers de ADN; se ha utilizado para Secuencia de la DNA, genoma a pie, y Huella de ADN.[35]
- PCR específica de metilación (MSP): desarrollado por Stephen Baylin y Jim Herman en la escuela de Medicina Johns Hopkins,[36] y se utiliza para detectar la metilación de islas CpG en la DNA genomic. ADN es primero tratado con bisulfito de sodio, que convierte unmethylated cytosine bases uracilo, que es reconocido por las cartillas de PCR como thymine. Dos PCRs se realizan en la DNA modificada, usando primer conjuntos idénticos, excepto en alguna islas de CpG en las secuencias de la cartilla. En estos momentos, un primer sistema reconoce metilado de la DNA con cytosines para amplificar la DNA, y un sistema reconoce la DNA con uracil o el thymine a amplificar unmethylated DNA. MSP utilizando qPCR puede realizarse también para obtener cuantitativos en lugar de información cualitativa sobre la metilación.
- Miniprimer PCR:: utiliza una polimerasa termoestable (S-Tbr) que puede extender de cebadores cortos ("smalligos") tan cortos como 9 o 10 nucleótidos. Este método permite PCR dirigidos a pequeñas regiones de enlace cartilla y se utiliza para amplificar secuencias de ADN conservadas, como el 16S (o 18S eukaryotic) gene del rRNA.[37]
- Amplificación Multiplex sonda de ligadura dependientes (MLPA): permite amplificar varios destinos con un par de la cartilla individual, evitando así las limitaciones de resolución de multiplex PCR (véase abajo).
- Múltiplex-PCR:: consta de varios conjuntos de primer dentro de una única mezcla PCR para producir amplicones de diferentes tamaños son específicos para diferentes secuencias de ADN. Dirigiéndose a varios genes a la vez, información adicional puede obtenerse de un funcionamiento de prueba solo que de lo contrario requeriría varias veces los reactivos y más tiempo para llevar a cabo. Temperaturas de recocido para cada uno de los conjuntos de la cartilla debe optimizarse para trabajar correctamente dentro de una sola reacción y tamaños de productos. Es decir, su longitud de base par debe ser lo suficientemente diferente como para formar grupos distintos cuando es visualizado por electroforesis en gel de.
- PCR de nanopartículas-asistida (nanoPCR):: En los últimos años se ha reportado que algunas nanopartículas (NPs) pueden mejorar la eficiencia de la PCR (así se llama el nanoPCR), y algunos incluso se desempeñan mejor que los originales potenciadores PCR. También fue encontrado que puntos cuánticos (QDs) pueden mejorar la eficiencia y especificidad de la PCR. Nanotubos de carbono de pared única (SWCNTs) y nanotubos de carbono multipared (ello) son eficaces en la mejora de la amplificación de la polimerización en cadena larga. Nanopolvos de carbono (PNC) informó ser capaces de mejorar la eficiencia de PCR repetida y polimerización en cadena larga. ZnO, TiO2, y Ag NPs también fueron encontrados para aumentar la producción PCR. Lo importante, ha indicado datos ya conocidos que NPs-metálico mantenido fidelidad amplificación aceptable. Dado que muchos NPs son capaces de mejorar la eficiencia de la PCR, es claro que es probable que gran potencial para el desarrollo de productos y mejoras de tecnología nanoPCR.[38][39]
- PCR anidada:: aumenta la especificidad de la amplificación de la DNA, reduciendo el fondo debido a la amplificación no específica de la DNA. Dos sistemas de cartillas se utilizan dos PCRs sucesivos. En la primera reacción, un par de cartillas se utiliza para generar productos de ADN, que además del objetivo, todavía puede consistir en fragmentos de ADN amplificados no específica. Los productos se utilizan luego en una segunda PCR con un conjunto de cartillas cuyos sitios de Unión son parcial o totalmente diferente y encuentra 3' de cada uno de los iniciadores utilizados en la primera reacción. Polimerización en cadena jerarquizada es a menudo más acertado en específicamente amplificar mucho fragmentos de DNA que PCR convencional, pero requiere más detallan conocimiento de las secuencias diana.
- Superposición-extensión PCR o Empalme por traslapo extensión (SOEing) : un ingeniería genética técnica que se utiliza para empalmar ADN de dos o más fragmentos contienen secuencias complementarias. Se utiliza para unir piezas de ADN que contienen genes, secuencias reguladoras o mutaciones; la técnica permite la creación de específicos y construcciones de ADN largo. También puede introducir supresiones, inserciones o mutaciones de punto en una secuencia de ADN.[40][41]
- PAN-AC:: utiliza las condiciones isotérmicas para la amplificación y puede ser utilizado en las células vivas.[42][43]
- PCR cuantitativa (qPCR): utilizado para medir la cantidad de una secuencia de destino (normalmente en tiempo real). Mide cuantitativamente cantidades a partir de RNA, DNA o cDNA. PCR cuantitativa se utiliza comúnmente para determinar si una secuencia de ADN está presente en una muestra y el número de sus copias en la muestra. PCR cuantitativa tiene un muy alto grado de precisión. Los métodos de PCR cuantitativos utilizan tintes fluorescentes, como el Sybr Green, EvaGreen o fluoróforo-que contengan ADN sondas, tales como TaqMan, para medir la cantidad de producto amplificado en tiempo real. También a veces se abrevia a RT-PCR (en tiempo real Polimerización en cadena) pero esta abreviatura debe utilizarse sólo para transcripción reversa PCR. qPCR es las contracciones adecuadas para PCR cuantitativa (PCR en tiempo real).
- Transcripción reversa PCR ()RT-PCR):: para amplificar el DNA del RNA. De la transcriptasa reversa transcribe del reverso RNA en cDNA, que luego es amplificado por PCR. RT-PCR es ampliamente utilizado en perfiles de expresión, para determinar la expresión de un gen o para identificar la secuencia de una transcripción del RNA, incluyendo sitios de inicio y terminación de transcripción. Si se conoce la secuencia genomic de la DNA de un gene, RT-PCR puede utilizarse para asignar la ubicación de exones y intrones en el gen. El extremo 5' de un gen (correspondiente a la página de inicio de transcripción) es típicamente identificado por RACE-PCR (Amplificación rápida del cDNA termina).
- Fase sólida PCR:: abarca significados múltiples, incluyendo Amplificación de Polonia (donde las colonias PCR se derivan en una matriz de gel, por ejemplo), PCR de puente[44] (cebadores son covalente a una superficie sólida de apoyo), PCR de fase sólida convencional (donde PCR asimétrico se aplica en presencia de sólido apoyo cojinete cartilla con secuencia de la coincidencia de uno de los primers acuosos) y PCR de fase sólida mejorada[45] (donde PCR convencional de fase sólida puede mejorarse mediante el empleo de alta Tm y cartilla de apoyo sólidos anidados con uso opcional de un térmico 'paso' a cebado apoyo sólido a favor).
- Suicidio PCR:: normalmente se usa en paleogenetics u otros estudios donde evitando falsos positivos y garantizar la especificidad del fragmento amplificado son la prioridad más alta. Originalmente fue descrito en un estudio para verificar la presencia del microbio Yersinia pestis en las muestras dentales obtenidas de tumbas del siglo XIV de personas supuestamente asesinadas por plaga durante la edad media Muerte negra epidemia.[46] El método prescribe el uso de cualquier combinación de cartilla sólo una vez en una polimerización en cadena (de ahí el término "suicidio"), que no deben nunca han sido utilizados en ningún control positivo reacción de PCR y los iniciadores siempre deben dirigir una región genómica no amplificada nunca antes en el laboratorio utilizando este o cualquier otro conjunto de cartillas. Esto asegura que ninguna DNA contaminante de reacciones de PCR anteriores está presente en el laboratorio, que de lo contrario puede generar falsos positivos.
- Asimétrico termal entrelazado (polimerización en cadenaCOLA-PCR):: para el aislamiento de una secuencia desconocida que flanquea una secuencia conocida. Dentro de la secuencia conocida, cola-PCR utiliza un par de cebadores anidado con diferentes temperaturas de recocido; una cartilla degenerada se utiliza para amplificar en la otra dirección de la secuencia desconocida.[47]
- Momento del aterrizaje PCR (PCR de descender): una variante de PCR que pretende reducir el fondo no específico bajando gradualmente la temperatura de recocido como progresa ciclo PCR. La temperatura del recocido en los ciclos iniciales es generalmente algunos grados (3 – 5 ° C) por encima de la Tm de los cebadores utilizados, mientras que en los ciclos posteriores, es unos pocos grados (3 – 5 ° C) por debajo del primer Tm. Las temperaturas más altas dan mayor especificidad para el atascamiento de la cartilla, y las temperaturas más bajas permiten una amplificación más eficiente de los productos específicos formados durante los ciclos iniciales.[48]
- Caminar rápido universal:: para caminar de genoma y huella genética usando un PCR más específica 'dos caras' de enfoques convencionales 'unilaterales' (con solamente una cartilla gene-específica y una cartilla general — que puede conducir a artefactual 'ruido')[49] en virtud de un mecanismo que implica la formación de la estructura de lazo. Derivados aerodinámico de UFW son carril de rabia (lariat-dependiente PCR anidado para la amplificación rápida de los extremos de ADN genómicos de),[50] 5' carrera LaNe[51] y 3' carril de la raza.[52]
Historia

Un documento de 1971 en la Diario de la Biología Molecular por Kjell Kleppe y compañeros de trabajo en el laboratorio de H. Gobind Khorana descrita por primera vez un método usando un ensayo enzimático para replicar una corta plantilla ADN con cebadores en vitro.[53] Sin embargo, esta manifestación temprana del principio básico de la PCR no recibió mucha atención en el momento, y la invención del reacción en cadena de la polimerasa en 1983 es generalmente acreditada a Kary Mullis.[54]

Cuando Mullis desarrolló la PCR en 1983, trabajaba Emeryville, California para CETUS Corporation, uno de los primeros Biotecnología empresas. Allí, él era responsable de sintetizar cadenas cortas de ADN. Mullis ha escrito que él concibió de PCR durante la navegación a lo largo de la Pacific Coast Highway una noche en su coche.[55] Estaba jugando en su mente con una nueva forma de analizar los cambios (mutaciones) en el ADN cuando se dio cuenta que él había inventado en su lugar un método de amplificación de cualquier región de ADN mediante ciclos repetidos de duplicación por polimerasa de la DNA. En Americano científico, Mullis resumió el procedimiento: "a partir de una sola molécula de ADN material genético, la PCR puede generar 100 billones de moléculas similares en una tarde. La reacción es fácil de ejecutar. Requiere no más que un tubo de ensayo, algunos reactivos simples y una fuente de calor."[56] Fue galardonado con el Premio Nobel de química en 1993 por su invento,[5] siete años después de que él y sus colegas a Cetus primero ponen su propuesta a la práctica. Sin embargo, se han mantenido algunas controversias sobre las contribuciones intelectuales y prácticas de otros científicos al trabajo de Mullis, y si él había sido el inventor único del principio PCR (véase abajo).
En el núcleo de la polimerización en cadena el método es el uso de un adecuado Polimerasa de la DNA capaces de soportar las altas temperaturas de > 90 ° C (194 ° F) requerida para la separación de las dos hebras de ADN en la Doble hélice de ADN después de cada replicación ciclo. Las polimerasas de ADN empleadas inicialmente para en vitro experimentos presagiando PCR fueron incapaces de soportar estas altas temperaturas.[3] Así que los primeros procedimientos para la replicación del ADN fueron muy ineficientes y tiempo consumiendo y requieren grandes cantidades de ADN polimerasa y manejo continuo durante todo el proceso.
El descubrimiento en 1976 de Taq polimerasa— una polimerasa de la DNA purificada a partir de la bacteria termófila, Aquaticus de Thermus, que naturalmente vive en ambientes calientes (50 a 80 ° C (122 a 176 ° F))[12] como aguas termales, allanó el camino para las mejoras dramáticas del método PCR. La polimerasa de la DNA aislada de T. aquaticus es estable a altas temperaturas, permaneciendo activa incluso después de la desnaturalización del ADN,[13] evitando la necesidad de Añadir nueva polimerasa de la DNA después de cada ciclo.[4] Esto permitió un proceso automatizado basado en el termociclador para amplificación de ADN.
Controversias sobre patentes
La técnica de PCR fue patentada por Kary Mullis y asignada a CETUS Corporation, donde Mullis trabajó cuando él inventó la técnica en 1983. El Taq enzima polimerasa también fue cubierto por las patentes. Ha habido varios juicios de alto perfil relacionado con la técnica, incluyendo un pleito fracasado traído por Du Pont. La compañía farmacéutica Hoffmann-La Roche compró las derechas a las patentes en 1992 y actualmente tiene los que todavía están protegidos.
A relacionados con la batalla de patente sobre la enzima polimerasa de Taq está todavía en curso en varias jurisdicciones alrededor del mundo entre Roche y Promega. Las discusiones legales han extendido más allá de la vida de las original PCR Taq polimerasa patentes y, que expiró el 28 de marzo de 2005.[57]
Referencias
- ^ Bartlett, J. M. S.; Stirling, D. (2003). "Una historia corta de la reacción en cadena de polimerasa". Protocolos de PCR. Métodos en Biología Molecular 226 (2ª ed.). págs. 3 – 6. doi:10.1385/1-59259-384-4:3. ISBN1-59259-384-4.
- ^ US4683195
- ^ a b Saiki, R.; Scharf, S.; Faloona, f el.; Mullis, K.; Horn, G.; Erlich, H.; Arnheim, N. (1985). "Amplificación enzimática de secuencias genómicas de beta-globina y análisis del sitio de restricción para el diagnóstico de anemia de células falciformes". Ciencia 230 (4732): 1350-1354. doi:10.1126/Science.2999980. PMID2999980.
- ^ a b Saiki, R.; Gelfand, D.; Stoffel, S.; Scharf, S.; Higuchi, R.; Horn, G.; Mullis, K.; Erlich, H. (1988). "Cartilla-dirigió amplificación enzimática de DNA con una polimerasa termoestable de la DNA". Ciencia 239 (4839): 487-491. doi:10.1126/Science.2448875. PMID2448875.
- ^ a b Kary Mullis Nobel Conferencia, 08 de diciembre de 1993
- ^ Cheng, S.; Fockler, C.; Barnes, W. M.; Higuchi, R. (1994). "Amplificación eficaz de blancos largas de insertos clonados y ADN genómico humano". Actas de la Academia Nacional de Ciencias 91 (12): 5695-5699. doi:10.1073/pnas.91.12.5695. PMC44063. PMID8202550.
- ^ Carr CA, SD de Moore (2012). Lucia, Alejandro, ed. "Robusta cuantificación de reacciones en cadena de polimerasa usando conexión global". PloS uno 7 (5): e37640. doi:10.1371/journal.pone.0037640. PMC3365123. PMID22701526.
- ^ a b José Sambrook y David W. Russel (2001). Clonación molecular: Un Manual de laboratorio (3ª Ed.). Cold Spring Harbor, N.Y.: Prensa de laboratorio Cold Spring Harbor. ISBN0-879-69576-5. Capítulo 8: amplificación In vitro de ADN por reacción en cadena de la polimerasa
- ^ Pavlov, A. R.; Pavlova, N. V.; Kozyavkin, S. A.; Slesarev, A. I. (2004). «Novedades en la optimización de termoestables polimerasas de la DNA para applications☆ eficaz». Tendencias en biotecnología 22 (5): 253 – 260. doi:10.1016/j.tibtech.2004.02.011. PMID15109812.
- ^ W Rychlik, Spencer WJ, Rhoads RE (1990). "Optimización de la temperatura de recocido para la amplificación de DNA in vitro". Res de ácidos Nucl 18 (21): 6409-6412. doi:10.1093/Nar/18.21.6409. PMC332522. PMID2243783.
- ^ a b Sharkey, J. D.; Scalice, R. E.; Christy, K. G.; Atwood, S. M.; Daiss, j. L. (1994). "Anticuerpos como interruptores termolábiles: activación de alta temperatura para la reacción en cadena de polimerasa". Bio/Technology 12 (5): 506-509. doi:10.1038/nbt0594-506.
- ^ a b Chien A, Edgar DB, Trela JM (1976). "Polimerasa del ácido desoxirribonucléico de la extrema termófila Thermus aquaticus". J Bacteriol 127 (3): 1550-1557. PMC232952. PMID8432.
- ^ a b Abogado, f el.; Stoffel, S.; Saiki, R.; Chang, S.; Landre, P.; Abramson, R.; Gelfand, D. (1993). "Expresión de alto nivel, purificación y caracterización enzimática de larga duración Thermus aquaticus DNA polimerasa y una forma truncada deficiente en 5' a 3' actividad exonucleasa." Aplicaciones y métodos de PCR 2 (4): 275 – 287. doi:10.1101/gr.2.4.275. PMID8324500.
- ^ Biología Campbell, 7ª edición
- ^ PCR a partir de plantillas de problemáticas. 22:1 p (2000) se centran.
- ^ Consejos útiles para PCR. Enfoque p.12 22:1 (2000).
- ^ Sarkar, G.; Kapelner, S.; Sommer, S. (1990). «Formamida puede mejorar considerablemente la especificidad de la PCR». Investigación de ácidos nucleicos 18 (24): 7465. doi:10.1093/Nar/18.24.7465. PMC332902. PMID2259646.
- ^ "PCR electrónica". NCBI-Centro Nacional para información biotecnológica. 13 de marzo 2012.
- ^ Pavlov AR, Pavlova NV, Kozyavkin SA, Slesarev AI (2006). "Polimerasas termoestables de la DNA para un amplio espectro de aplicaciones: comparación de un TopoTaq híbrido robusto a otras enzimas". En J. Kieleczawa DNA secuencia II: Optimización de la preparación y limpieza. Jones y Bartlett. págs. 241-257. ISBN0-7637-3383-0.
- ^ "Síntesis químicas, secuenciación y amplificación de ADN (notas de clase sobre MBB/BIO 343)". Universidad Estatal de Arizona. 2007-10-29.
- ^ a b c CAI, H; Caswell JL; Prescott JF (marzo de 2014). «Nonculture técnicas moleculares para el diagnóstico de enfermedades bacterianas en animales: una perspectiva de diagnóstico de laboratorio». Patología Veterinaria 51 (2): 341-350. doi:10.1177/0300985813511132. PMID24569613.
- ^ Salis AD (2009). "Aplicaciones en microbiología clínica". PCR en tiempo real: Tecnología actual y aplicaciones. Prensa académica de Caister. ISBN978-1-904455-39-4.
- ^ LILIT Garibyan y Nidhi A (2013). "Reacción en cadena de polimerasa". Revista de Dermatología investigativa 133. doi:10.1038/JID.2013.1.
- ^ CR de Newton, A Graham, Heptinstall LE, Powell SJ, veranos C, Kalsheker N, Smith JC y Markham AF (1989). "Análisis de cualquier punto de mutación en el ADN. El sistema de mutación refractario de amplificación (brazos)". Investigación de ácidos nucleicos 17 (7): 2503-2516. doi:10.1093/Nar/17.7.2503. PMC317639. PMID2785681.
- ^ Stemmer WP, Crameri A, Ha KD, Brennan TM, Heyneker HL (1995). "Montaje single-step de un gen y el plásmido todo de grandes números de oligodeoxyribonucleotides". Gene 164 (1): 49 – 53. doi:10.1016/0378-1119 (95) 00511-4. PMID7590320.
- ^ Innis MA, Myambo KB, Gelfand DH, ceja MA. (1988). "Secuencia de la DNA con Thermus aquaticus polimerasa de la DNA y la secuencia directa del DNA amplificado de la reacción en cadena de polimerasa". Proc Natl Acad Sci USA 85 (24): 9436-4940. doi:10.1073/pnas.85.24.9436. PMC282767. PMID3200828.
- ^ KE de Pierce y método LJ (2007). Reacción en cadena de polimerasa Linear-después de-la-exponencial y tecnologías afines estrategias de detección en tiempo real para el diagnóstico rápido y fiable de las células. Métodos Mol Med. Métodos moleculares medicina ™ 132:: 65 – 85. doi:10.1007/978-1-59745-298-4_7. ISBN978-1-58829-578-1. PMID17876077.
- ^ Schwartz JJ, Lee C, Shendure J. (2012). "Síntesis precisa gene con etiqueta indica recuperación de moléculas de ADN de secuencia verificada". Métodos de naturaleza 9 (9): 913-915. doi:10.1038/nmeth.2137. PMC3433648. PMID22886093.
- ^ Vincent, Myriam, Xu, Yan y Kong, Huimin (2004). "Amplificación del ADN isotérmica Helicase-dependiente". Informes EMBO 5 (8): 795-800. doi:10.1038/sj.embor.7400200. PMC1249482. PMID15247927.
- ^ P. Chou, Russell M., D.E. abedul, Raymond J. y W. Bloch (1992). "Prevención de la dimerización de mal cebado y cartilla de pre-PCR mejora amplificaciones de bajo número de copias". Investigación de ácidos nucleicos 20 (7): 1717-1723. doi:10.1093/Nar/20.7.1717. PMC312262. PMID1579465.
- ^ Kellogg s D E, Rybalkin I, Chen S, Mukhamedova N, T Vlasik, Siebert P D, Chenchik A. 1994. Anticuerpo TaqStart: hot start PCR facilitada por anticuerpos monoclonales neutralizantes dirigidos contra DNA de Taq polimerasa. BioTechniques 16 (6): 1134-1137Kellogg, DE; Rybalkin, I; Chen, S; Mukhamedova, N; Vlasik, T; Siebert, PD; Chenchik, (1994). "Anticuerpos TaqStart:" comienzo caliente "Facilitado por un anticuerpo monoclonal neutralizante dirigido contra la polimerasa de la DNA de Taq PCR". BioTechniques 16 (6): 1134-7. PMID8074881.
- ^ San Millan RM, Martínez Ballesteros I, Rementeria A, Garaizar J, Bikandi J (2013). «Ejercicio en línea para el diseño y simulación de experimentos de PCR y PCR-RFLP». BMC Research Notes 6:: 513. doi:10.1186/1756-0500-6-513. PMID24314313.
- ^ E. Zietkiewicz, Rafalski A. y D. Labuda (1994). "Repita el genoma huellas dactilares por secuencia simple (SSR)-anclado la amplificación la reacción en cadena de polimerasa". Genomics 20 (2): 176-83. doi:10.1006/geno.1994.1151. PMID8020964.
- ^ Ochman H, como Gerber, Hartl DL (1988). "Aplicaciones genéticas de una reacción en cadena de polimerasa inversa". Genética 120 (3): 621-623. PMC1203539. PMID2852134.
- ^ Mueller PR, sería B (1988). "En vivo huella de un potenciador específico del músculo por la ligadura mediada por PCR". Ciencia 246 (4931): 780-786. doi:10.1126/Science.2814500. PMID2814500.
- ^ Herman JG, Graff JR, S Myöhänen, Nelkin BD, Baylin SB (1996). "PCR específica de metilación: un nuevo ensayo PCR para el estado de metilación de islas CpG". Proc Natl Acad Sci USA 93 (13): 9821-9826. doi:10.1073/pnas.93.18.9821. PMC38513. PMID8790415.
- ^ Isenbarger TA Finney M, Ríos Velázquez C, Handelsman J, Ruvkun G (2008). "Miniprimer PCR, una nueva lente para ver el mundo microbiano". Microbiología aplicada y ambiental 74 (3): 840 – 9. doi:10.1128/AEM.01933-07. PMC2227730. PMID18083877.
- ^ Cenchao Shen Yang Wenjuan, ahora Ji, Hisaji Maki, Anjie Dong, Zhizhou Zhang (2009). "Observación de NanoPCR: diferentes niveles de fidelidad de replicación del ADN en reacciones en cadena de polimerasa mejorado de nanopartículas". Nanotecnología 20:: 455103. doi:10.1088/0957-4484/20/45/455103.
- ^ Shen, Cenchao (2013). Un resumen de la tecnología de reacción en cadena de polimerasa Nanoparticle‐Assisted. Estados Unidos: Wiley-Blackwell Publishing Ltd. pp. 97 – 106.
- ^ Horton RM, caza HD, Ho SN, JK Pullen, Pease LR (1989). "Ingeniería genes híbridos sin el uso de enzimas de restricción: gen de empalme por traslapo exten-sion". Gene 77 (1): 61 – 68. doi:10.1016/0378-1119 (89) 90359-4. PMID2744488.
- ^ Moller, Simon (2006). PCR (CONCEPTOS BÁSICOS). Estados Unidos: Taylor & Francis Group. p. 144.
- ^ David, f el. y Turlotte, E., (1998). "Un método de amplificación isotérmica". C.R.Acad. Sci París, Ciencias de la vida 321 (1): 909-914. doi:10.1016/S0764-4469 (99) 5 80005.
- ^ Fabrice David (septiembre – octubre 2002). "Signo les propriétés topologiques de l'ADN: une nouvelle arme contre les agentes pathogènes" (PDF) 92. Fusión.(en francés)
- ^ Bing, H. D., C. Boles, F. N. Rehman, M. Audeh, M. Belmarsh, B. Kelley y C. p. Adams. (1996). "Puente de amplificación: un sistema de PCR en fase sólida para la amplificación y detección de diferencias alélicas en los genes de copia única". Actas de identidad genética, VII Simposio Internacional sobre identificación humana.
- ^ Khan Z, Poetter K, Parque DJ (2008). «Fase sólida mejorada PCR: mecanismos para aumentar el oscurecimiento por cartillas de apoyo sólido ". Bioquímica analítica 375 (2): 391-393. doi:10.1016/j.AB.2008.01.021. PMID18267099.
- ^ Raoult, D; Aboudharam G; Crubezy E; G Larrouy; Ludes de B; Drancourt M (2000-11-07). "Identificación molecular por suicidio" PCR"de pestis de Yersinia como el agente de la muerte negra medieval". Estados Unidos el proc. nacional Acad. SCI. 97 (23): 12800-12803. doi:10.1073/pnas.220225197. ISSN0027-8424. PMC18844. PMID11058154.
- ^ Y.G. Liu y R. F. Whittier (1995). «Asimétrico termal entrelazado PCR: automatizable amplificación y secuenciación del inserto fin fragmentos de P1 y YAC clones para caminar del cromosoma ". Genomics 25 (3): 674 – 81. doi:10.1016/0888-7543 (95) 80010-J. PMID7759102.
- ^ Don Cox PT, Wainwright BJ, RH, panadero K, Mattick JS (1991). "'Touchdown' PCR para eludir falsa cebado durante la amplificación del gen ". Res de ácidos Nucl 19 (14): 4008. doi:10.1093/Nar/19.14.4008. PMC328507. PMID1861999.
- ^ Myrick KV, Gelbart WM (2002). "Universal rápido caminar de determinación directa y versátil de que flanquean la secuencia". Gene 284 (1-2): 125 – 131. doi:10.1016/S0378-1119 (02) 00384-0. PMID11891053.
- ^ Parque de DJ electrónico diario de la biotecnología (en línea). 15 de agosto de 2005, vol. 8, núm. 2
- ^ Parque DJ (2005). "Un nuevo 5' terminal murino GAPDH exón identificada mediante 5' carril de la raza". Biotecnología molecular 29 (1): 39 – 46. doi:10.1385 / MB:29:1:39. PMID15668518.
- ^ Parque DJ (2004). «3' carril de la raza: un método PCR completamente anidado simple y rápido para determinar la secuencia del cDNA 3'-terminal ". Biotechniques 36 (4): 586-588, 590. PMID15088375.
- ^ K Kleppe, Ohtsuka E, Kleppe R, Molineux I, Khorana hectogramo (1971). "Estudios de polinucleótidos. Repeticiones de XCVI. reparación de la DNA sintético corto de como catalizada por las polimerasas de la DNA". J. análizar Biol 56 (2): 341-361. doi:10.1016/0022-2836 (71) 90469-4. PMID4927950.
- ^ Rabinow, Paul (1996). Fabricación de PCR: Una historia de la biotecnología. Chicago: University of Chicago Press. ISBN0-226-70146-8.
- ^ Mullis, Kary (1998). Bailando desnuda en el campo de la mente. Nueva York: Pantheon Books. ISBN0-679-44255-3.
- ^ Mullis, Kary (1990). "El origen inusual de la reacción en cadena de polimerasa". Americano científico 262 (4): 56 – 61, 64-5. doi:10.1038/scientificamerican0490-56. PMID2315679.
- ^ Consejos sobre cómo sobrevivir el Taq guerrea ¶2: Noticias de la ingeniería genética de GEN Biobusiness Channel: artículo. 01 de mayo de 2006 (Vol. 26, no. 9).
Enlaces externos
| Recursos de la biblioteca Acerca de Reacción en cadena de polimerasa |
|
| Campos comunes de Wikimedia tiene medios relacionados con Reacción en cadena de polimerasa. |
- Una guía a las tecnologías de PCR SelectScience
- OpenPCR Proyecto de código abierto PCR thermalcycler
- Patente de los E.E.U.U. para PCR
- Paso por la animación de PCR – Laboratorio de Cold Spring Harbor
- OpenWetWare
- ¿Cuál es el efecto de meseta PCR? YouTube tutorial video
- GeneWarrior En línea herramienta de diseño de la cartilla de la polimerización en cadena
- Historia de la reacción en cadena de polimerasa desde el Archivos de institución Smithsonian
- modelos 3D de equipos de PCR para la impresión 3D en thingiverse.com
- Ejercicio de equipo. Diseño de experimentos PCR y PCR-RFLP
|
||||||
|