Neoplasma
| Neoplasma | |
|---|---|
| Clasificación y recursos externos | |

Colectomía muestras que contengan una neoplasia maligna, es decir un invasor carcinoma colorrectal (el tumor similar al cráter, rojizo y de formado irregular)
|
|
| CIE-10 | C00-D48 |
| CIE-9 | 140-239.99 |
| DiseasesDB | 28841 |
| MedlinePlus | 001310. |
| Malla | D009369 |
Neoplasma (a partir de Griego antiguo ΝΕΟ- Neo- "nuevo" y πλάσμα plasma "la formación, creación"), también comúnmente conocido como un tumor o tumor,[1] es un crecimiento anormal del tejido.[2] Este crecimiento anormal generalmente pero no siempre forma una masa.[3]
El Organización Mundial de la salud clasifica neoplasias en cuatro grupos principales: Neoplasias benignas, Neoplasias in situ, Neoplasias malignasy los tumores de comportamiento incierto o desconocido.[4] Neoplasia maligna es un cáncer.
Antes de crecimiento anormal (conocido como neoplasia), células a menudo se someten a un patrón anormal de crecimiento, tales como metaplasia o displasia.[5] Sin embargo, la metaplasia o displasia no siempre progresa a la neoplasia. El crecimiento de las células neoplásicas excede y no está coordinado con la de los tejidos normales alrededor de él. El crecimiento continúa de la misma manera excesiva incluso después de la cesación de los estímulos. Generalmente causa una protuberancia o tumor.
En la medicina moderna, el término tumor significa un neoplasma que se ha formado un bulto. En el pasado, el término tumor se utilizó de forma diferente,[citación necesitada] refiriéndose a un trozo de cualquier causa. Algunas neoplasias no causan una protuberancia.
Contenido
- 1 Tipos
- 2 Definición
- 2.1 Clonalidad
- 2.2 Neoplasia vs tumor
- 3 Causas
- 4 Neoplasias malignas
- 4.1 Daños en el ADN
- 4.2 Defectos del campo
- 4.3 Inestabilidad genómica
- 5 Etimología
- 6 Véase también
- 7 Referencias
Tipos
Un neoplasma puede ser benigno, potencialmente malignas (cancerosas previa), o (maligno)cáncer).[6]
- Neoplasias benignas incluyen fibromas uterinos y nevos melanocíticos (lunares de la piel). Son circunscritos y localizadas y no se transforme en cáncer.[5]
- Neoplasias potencialmente malignas incluyen carcinoma in situ. No invadir y destruir pero, con el tiempo, se transformará en un cáncer.
- Los tumores malignos son comúnmente llamados cáncer. Ellos invaden y destruyen el tejido que lo rodea, pueden forma metástasis y finalmente mata al anfitrión.
- Neoplasma secundario se refiere a cualquiera de una clase de tumor canceroso que es una rama de metástasis de un tumor primario, o un tumor aparentemente no relacionado que aumenta en frecuencia siguiendo ciertos tratamientos contra el cáncer tales como quimioterapia o radioterapia.
Definición
| -plasia y - trofeo |
|---|
|
|
Debido a la neoplasia incluye enfermedades muy diferentes, es difícil encontrar una definición que abarque todo.[7] La definición de los británicos oncólogo R.A. Willis es ampliamente citado: "una neoplasia es una masa anormal de tejido, el crecimiento que excede y está descoordinado con el de los tejidos normales y persiste en la misma manera excesiva después de la cesación del estímulo que evoca el cambio".[8] Esta definición es criticada debido a algunas neoplasias, como nevos, no son progresivos.
Clonalidad
Los tumores neoplásicos a menudo contienen más de un tipo de célula, pero su iniciación y crecimiento continuo es generalmente dependiente de una sola población de células neoplásicas. Se presume que estas células son clonal – es decir, descienden de una célula del progenitor único.
A veces, las células neoplásticas todos llevan el mismo genética o Epigenética anomalía que se convierte en pruebas de clonalidad. Para las neoplasias linfoides, e.g. linfoma y leucemia, clonalidad es probada por la amplificación de un simple cambio de su inmunoglobulina gen (para Célula de b las lesiones) o Receptor de células T gen (para Célula de t lesiones). La demostración de clonalidad es considerada necesario identificar una proliferación de células linfoides como neoplásicas.[9]
Es tentador definir Neoplasias como clonales proliferaciones celulares pero la demostración de clonalidad no es siempre posible. Por lo tanto, no es necesario clonalidad en la definición de la neoplasia.
Neoplasia vs tumor
Tumor (En latín hinchazónuno de los signos cardinales de la inflamación) originalmente significaba cualquier forma de hinchazón, neoplásicas o no. Español actual, sin embargo, usos médicos y no médicos, tumor como sinónimo de neoplasma.[10]
Algunas neoplasias no forman un tumor. Estos incluyen leucemia y la mayoría de las formas de carcinoma in situ.
Un tumor (Inglés americano) o (tumor)Inglés británico) es comúnmente utilizado como un sinónimo para un neoplasma[11] (un sólido o llenos de líquido quístico lesión puede o no puede estar formado por un crecimiento anormal de neoplásicas las células) que aparece agrandado en tamaño.Tumor No es sinónimo de cáncer. Mientras que el cáncer es por definición maligno, un tumor puede ser benigno, premalignas, o maligno.
Los términos "masa" y "nódulo" se utilizan a menudo sinónimo con el "tumor". En términos generales, sin embargo, el término "tumor" se utiliza genéricamente, sin hacer referencia al tamaño físico de la lesión. Más concretamente, el término "masa" se utiliza a menudo cuando la lesión tiene un diámetro máximo de al menos 20 milímetros (mm) en la dirección más grande, mientras que el término"nódulo"se utiliza generalmente cuando el tamaño de la lesión es menos de 20 mm en su mayor dimensión (25,4 mm = 1 pulgada).
Causas
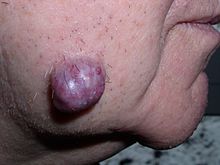

Una neoplasia puede ser causada por una proliferación anormal de tejidos, que puede ser causada por genética mutaciones. No todos los tipos de Neoplasias causan un crecimiento excesivo con tumores del tejido, sin embargo (tales como leucemia o carcinoma in situ).
Recientemente, se ha estudiado el crecimiento del tumor usando matemáticas y continuo de la mecánica. Los tumores vasculares se miran así como ser las amalgamas de un esqueleto sólido formado por células pegajosas y un líquido orgánico llenando los espacios en los que las células pueden crecer.[12] Bajo este tipo de modelo, cepas y tensiones mecánicas pueden ser tratadas y su influencia en el crecimiento del tumor y el tejido circundante y vasculatura dilucidado. Recientes hallazgos de los experimentos que utilizan este modelo muestran que el crecimiento activo del tumor está restringido a los bordes exteriores del tumor, y que la rigidez del tejido normal subyacente inhibe así como el crecimiento del tumor.[13]
Condiciones benignas que son No asociada a una proliferación anormal de tejido (tales como quistes sebáceos) también puede presentarse como tumores, sin embargo, pero no que ningún potencial maligno. Quistes de mama (como ocurre comúnmente durante el embarazo y en otras ocasiones) son otro ejemplo, como otro encapsulado inflamiento glandular (tiroides, glándula suprarrenal, páncreas).
Los hematomas encapsulados, encapsulado tejido necrótico (de la picadura de un insecto, cuerpo extraño u otro mecanismo nocivo), queloides (overgrowths discretas de tejido cicatricial) y granulomas también puede presentarse como tumores.
Discretas ampliaciones localizadas de las estructuras normales (uréteres, vasos sanguíneos, los conductos biliares intrahepáticos o extrahepáticos, inclusiones pulmonares, o duplicaciones gastrointestinales) debido a la obstrucción de la salida o estrechamientos o conexiones anormales, también puede presentarse como un tumor. Los ejemplos son fístulas arteriovenosas o aneurismas (con o sin trombosis), fístulas biliares o aneurismas, colangitis esclerosante primaria, quistes cisticercosis o de la hidátide, duplicaciones intestinales y pulmonares inclusiones como se ha visto con fibrosis quística. Puede ser peligroso para biopsia un número de tipos de tumor en el que la fuga de sus contenidos potencialmente podría ser catastrófica. Cuando estos tipos de tumores son modalidades encontradas, diagnósticos como ultrasonido, exploraciones de CT, MRI, angiogramas y medicina nuclear son la biopsia empleadas antes (o durante) o exploración/supresión quirúrgica en un intento por evitar tales complicaciones severas.
La naturaleza de un tumor es determinada por la proyección de imagen, por la exploración quirúrgica, o por un patólogo después de la examinación del tejido de una biopsia o una espécimen quirúrgico.
Neoplasias malignas
Daños en el ADN

Daños en el ADN se consideran la principal causa subyacente de neoplasias malignas conocido como cánceres.[14][15] Su papel central en la progresión del cáncer se ilustra en la figura en esta sección, en el recuadro de la parte superior. (Las características centrales de daños en el ADN, Epigenética alteraciones y deficiente reparación del ADN en la progresión del cáncer aparecen en rojo.) Daños en el ADN son muy común. Naturalmente que ocurren daños del ADN (en su mayoría debido al metabolismo celular y las propiedades del ADN en agua a temperatura corporal) ocurren a un ritmo de más de 60.000 nuevos daños, en promedio, por la célula humana, por día[14] [Consulte también el artículo Daños en el ADN (natural) ]. Daños adicionales de ADN pueden derivarse de la exposición a exógeno agentes. Causas de humo de tabaco aumentadas exógeno Daños en el ADN y estos daños de ADN son la causa probable de cáncer de pulmón debido al tabaquismo.[16] La luz UV de la radiación solar provoca daños en el ADN que es importante en el melanoma.[17] Infección por Helicobacter pylori produce altos niveles de especies reactivas de oxígeno dañan el ADN y contribuye al cáncer gástrico.[18] Ácidos biliares, en niveles elevados en los dos puntos de los seres humanos comen una dieta alta en grasas, también causan daños en el ADN y contribuir al cáncer de colon.[19] Katsurano et al. indicaron que los macrófagos y neutrófilos en un epitelio colon inflamado son la fuente de especies reactivas de oxígeno causando los daños del ADN que inician la tumorigénesis colónica.[20] En las cajas en la parte superior de la figura en esta sección se indican algunas fuentes de daño en el ADN.
Individuos con una mutación de línea germen que causa deficiencia en cualquiera de los genes de reparación de ADN 34 (ver artículo Trastorno de deficiencias en la reparación de ADN) corren un mayor riesgo de cáncer. Algunas mutaciones germinales de línea en el ADN reparacion genes causa arriba al azar 100% toda la vida del cáncer (por ejemplo, las mutaciones de p53).[21] Estas mutaciones de línea del germen se indican en una caja en la parte izquierda de la figura con una flecha que indica su contribución a la deficiencia de la reparación del ADN.
Alrededor del 70% de los tumores malignos no tienen ningún componente hereditario y son llamado "cánceres esporádicos".[22] Sólo una minoría de los cánceres esporádicos tienen una deficiencia en el ADN de reparación debido a la mutación en un gen de reparación del ADN. Sin embargo, la mayoría de los cánceres esporádicos tienen deficiencia en la reparación del ADN debido a Epigenética las alteraciones que reducen o silenciar ADN reparacion la expresión génica. Por ejemplo, para los cánceres colorrectales secuenciales 113, sólo cuatro tenían una mutación sin sentido en el gen de reparación del ADN MGMT, mientras que la mayoría había reducido la expresión MGMT debido a la metilación de la región del promotor MGMT (una alteración epigenética).[23] Cinco informes presentar evidencia que entre 40% y 90% de los cánceres colorrectales han reducido la expresión MGMT debido a la metilación de la región del promotor MGMT.[24][25][26][27][28]
Del mismo modo, fuera 119 casos de incompatibilidad reparaciones deficientes de los cánceres colorrectales que carecían de ADN reparacion la expresión génica PMS2, PMS2 era deficiente en 6 debido a las mutaciones en el gene PMS2, mientras que en 103 casos PMS2 expresión era deficiente porque su socio emparejamiento MLH1 fue reprimida por metilación del promotor (proteína PMS2 es inestable en la ausencia de MLH1).[29] En los otros 10 casos, pérdida de expresión PMS2 era probablemente debido a la epigenética sobreexpresión de los microARN, miR-155, que abajo-regula MLH1.[30]
En otros ejemplos [tabulados en el artículo Epigenética (ver sección "ADN" reparar epigenética en cáncer)], se encontraron defectos epigenéticos en frecuencias de entre 13% y 100% para los genes de reparación del ADN BRCA1, WRN, FANCB, FANCFMGMT, MLH1, MSH2, MSH4, ERCC1, XPF, NEIL1 y ATM. Estos defectos epigenéticos se produjeron en varios tipos de cáncer (mama, ovario, colon y cabeza y cuello). Dos o tres deficiencias en expresión de ERCC1, XPF o PMS2 ocurren simultáneamente en la mayoría de los cánceres de colon 49 evaluados por Facista et al.[31] En una caja central en el tercer nivel de la parte superior de la figura en esta sección se muestran alteraciones causando la menor expresión de los genes de reparación de ADN, y la consecuente deficiencia de reparación de ADN se muestra en el cuarto nivel epigenético.
Cuando se reduce la expresión de los genes de reparación de ADN, ADN daños se acumulan en las células en un más alto que el nivel normal y estos daños exceso causan frecuencias crecientes de mutación o epimutation. Las tasas de mutación aumentan fuertemente en las células defectuosas en Reparación del ADN desajuste[32][33] o en homologous recombinational reparación (HRR).[34]
Durante la reparación de roturas de doble hebra de DNA, o reparación de otros daños del ADN, pueden causar sitios despejados incompleto de reparación Epigenética silenciamiento génico.[35][36] Deficiencias de reparación de ADN (nivel 4 en la figura) causan daños crecientes de ADN (nivel 5 en la figura) que resultan en mayores mutaciones somáticas y alteraciones epigenéticas (nivel 6 en la figura).
Campo defectos, tejido normal que aparece con múltiples alteraciones (y discuten en la sección siguiente), son los precursores comunes de desarrollo del clon desordenado e inapropiadamente proliferación de tejido en una neoplasia maligna. Tales defectos de campo (segundo nivel de la parte inferior de la figura) puede tener múltiples mutaciones y alteraciones epigenéticas.
Una vez que se ha formado un cáncer, usualmente tiene inestabilidad genómica. Esta inestabilidad es probable debido a la reducida la reparación del ADN o excesivo daño en el ADN. Debido a esa inestabilidad, el cáncer continúa evolucionando y producir clones de sub. Por ejemplo, un cáncer renal, muestreado en 9 zonas, tenía 40 mutaciones ubicuas, demostrando heterogeneidad del tumor (es decir, presente en todas las áreas del cáncer), 59 mutaciones compartieron por algunos (pero no todas las áreas), y 29 mutaciones "privadas" sólo presentan en una de las áreas del cáncer.[37]
Defectos del campo

Varios otros términos se han utilizado para describir esto fenómenoincluyendo "efecto de campo","cancerization del campo"y"campo carcinogénesis". El término "cancerization del campo" primero fue utilizado en 1953 para describir una zona o "campo" del epitelio que ha sido precondicionado por (en aquel momento) en gran parte desconocido los procesos con el fin de lo predisponen al desarrollo de cáncer.[38] Desde entonces, han utilizado los términos "cancerization del campo" y "defecto de campo" para describir premalignas del tejido en que están probables que surjan nuevos cánceres.
Defectos del campo son importantes en la progresión del cáncer.[39][40] Sin embargo, en la mayoría cancer research, señaló Rubin[41] "La inmensa mayoría de los estudios de investigación del cáncer se ha hecho en tumores bien definidos en vivo o en focos neoplásicos discretos in vitro. Sin embargo, hay evidencia que más del 80% de las mutaciones somáticas encontramos mutator fenotipo humano los tumores colorrectales ocurren antes del inicio de la expansión clonal terminal.[42] Del mismo modo, Vogelstein et al.[43] señalan que más de la mitad de las mutaciones somáticas identificadas en los tumores se produjo en una fase pre-neoplásicas (en un defecto del campo), durante el crecimiento de células aparentemente normales. Asimismo, las alteraciones epigenéticas presentes en los tumores pueden haber ocurrido en defectos pre-neoplásicas del campo.
Una vista ampliada de efecto de campo se ha denominado "efecto de campo etiológico", que abarca no sólo patológicos y moleculares cambios en las células pre-neoplásicas sino también las influencias de factores exógenos ambientales y cambios moleculares en el local microambiente en evolución neoplásica de iniciación tumoral a muerte paciente.[44]
En el colon, un defecto del campo probablemente surge por la selección natural de una célula mutante o epigenéticamente alterada entre las células en la base de uno de los criptas intestinales en el interior superficial del colon. Una célula madre mutante o epigenéticamente alterada puede sustituir las otras células cercanas por la selección natural. Por lo tanto, puede surgir un parche de tejido anormal. La figura en esta sección incluye una foto de un segmento recién resecado y abierto a lo largo del colon mostrando un cáncer de colon y cuatro pólipos. Por debajo de la foto hay un diagrama esquemático de cómo un parche grande de células mutantes o epigenéticamente alterados puede haberse formado, se muestra por la amplia zona de amarillo en el diagrama. Dentro de este primer gran parche en el diagrama (un gran clon de células), una segunda mutación o alteración Epigenética puede ocurrir para que una determinada célula madre adquiere una ventaja en comparación con otras células dentro del parche y puede expandir esta alterado la célula de vástago clonalmente formando un parche secundario, o sub clon, dentro de la revisión original. Esto se indica en el diagrama por cuatro pequeños parches de diferentes colores dentro de la zona original amarilla grande. Dentro de estos nuevos parches (sub clones), el proceso puede repetirse varias veces, indicado por las manchas todavía más pequeñas dentro de los cuatro parches secundarios (con diferentes colores en el diagrama) que se expanden clonalmente, hasta que las células madre que generan o bien pequeños pólipos o bien un tumor maligno (cáncer) se presentan. En la foto, un defecto del campo aparente en este segmento de un colon ha generado cuatro pólipos (marcados con el tamaño de los pólipos, 6mm, 5mm y dos de 3mm y un cáncer unos 3 centímetros a través de su dimensión más larga). Estos tumores también se indican, en el diagrama debajo de la foto, por 4 círculos tan pequeños (pólipos) y un área más grande rojo (cáncer). El cáncer en la foto se produjo en la zona del ciego del colon, donde el colon se une con el intestino (etiquetado) y donde el apéndice se produce (etiqueta). La grasa en la foto es externa a la pared exterior del colon. En el segmento de colon que se muestra aquí, el colon se cortó abierto longitudinalmente para exponer la superficie interna del colon y para mostrar el cáncer y pólipos que ocurren dentro de la guarnición epitelial interior del colon.
Si el general del proceso por el cual esporádicos surgen de los cánceres de colon es la formación de un clon pre-neoplásicas que se propaga por la selección natural, seguida por la formación de clones sub internas dentro del clon inicial y sub-sub-clones dentro de aquellos, entonces los cánceres de colon generalmente deben estar asociados con y ser precedidos, campos de anormalidad creciente que refleja la sucesión de acontecimientos premalignas. La región más extensa de la anormalidad (el área irregular amarillo ultraperiférica en el diagrama) reflejaría el evento más temprano en la formación de una neoplasia maligna.
En la evaluación experimental de deficiencias específicas de reparación de ADN en casos de cáncer, muchas deficiencias específicas de reparación de ADN mostraron también ocurren en los defectos del campo circundante esos cánceres. La mesa, a continuación, da ejemplos para que la deficiencia de reparación del ADN en un cáncer fue demostrada para ser causado por una alteración epigenética y algo bajas frecuencias con que el mismo causó epigenéticamente deficiencia de reparación de ADN fue encontrado en el defecto del campo circundante.
| Cáncer | Gene | Frecuencia en el cáncer | Frecuencia en defecto del campo | Ref. |
|---|---|---|---|---|
| Colorrectal | MGMT | 46% | 34% | [24] |
| Colorrectal | MGMT | 47% | 11% | [26] |
| Colorrectal | MGMT | 70% | 60% | [45] |
| Colorrectal | MSH2 | 13% | 5% | [26] |
| Colorrectal | ERCC1 | 100% | 40% | [31] |
| Colorrectal | PMS2 | 88% | 50% | [31] |
| Colorrectal | XPF | 55% | 40% | [31] |
| Cabeza y cuello | MGMT | 54% | 38% | [46] |
| Cabeza y cuello | MLH1 | 33% | 25% | [47] |
| Cabeza y cuello | MLH1 | 31% | 20% | [48] |
| Estómago | MGMT | 88% | 78% | [49] |
| Estómago | MLH1 | 73% | 20% | [50] |
| Esófago | MLH1 | 77% - 100% | 23-79% | [51] |
Algunos de los pequeños pólipos en el defecto del campo que se muestra en la foto del segmento de colon abierto pueden ser Neoplasias relativamente benignos. De pólipos de menos de 10mm de tamaño, encontrado durante la colonoscopia y seguido con repetición colonoscopias durante 3 años, 25% eran sin cambios en tamaño, 35% regresan o se redujo en tamaño mientras que el 40% creció en tamaño.[52]
Inestabilidad genómica
Los cánceres son conocidos para exhibir inestabilidad genómica o un fenotipo mutador.[53] El proteína ADN dentro del núcleo es aproximadamente 1.5% del ADN genómico total.[54] Dentro de esta proteína ADN (llamado el exoma), un tipo de cáncer de seno o colon promedio puede tener unos 60 a 70 proteína alterar las mutaciones, de las cuales aproximadamente 3 o 4 pueden ser mutaciones "driver", y los restantes pueden ser mutaciones «pasajero»[43] Sin embargo, la secuencia de mutaciones en el genoma completo (incluyendo el número promedio de ADN regiones no codificantes de proteínas) dentro de una mama muestra de tejido de cáncer es de unos 20.000.[55] En una muestra de tejido de melanoma promedio (donde los melanomas tienen una mayor exoma frecuencia de la mutación[43]) el número de mutaciones de la secuencia del DNA es aproximadamente 80.000.[56] Esto se compara con la frecuencia de mutación muy baja de aproximadamente 70 nuevas mutaciones en el genoma entero entre generaciones (madre a hijo) en seres humanos.[57][58]
La alta frecuencia de mutaciones en las secuencias nucleotídicas total dentro de los cánceres sugieren que a menudo una alteración temprana en los defectos del campo dando lugar a un cáncer (e.g. área amarilla en el diagrama en esta sección) es una deficiencia en la reparación del ADN. Los defectos de campo grande alrededor de los cánceres de colon (extendiendo a unos 10 cm a cada lado de un cáncer) fueron demostrados por Facista et al.[31] con frecuencia tener defectos epigenéticos en 2 o 3 (proteínas de reparación del ADNERCC1XPF o PMS2) en el área entera del defecto del campo. Las deficiencias en el ADN de reparación causa aumentada las tasas de mutación.[32][33][34] Una deficiencia en la reparación del ADN, sí mismo, puede permitir ADN daños a acumular y error-prone síntesis de translesion más allá de algunos de los daños que pueden dar lugar a mutaciones. Además, reparación defectuosa de estos daños acumulados de ADN puede dar lugar a epimutations. Estas nuevas mutaciones o epimutations puede proporcionar una ventaja proliferativa, generando un defecto del campo. Aunque las mutaciones/epimutations en los genes de reparación de ADN no lo hacen, ellos mismos, aportan una ventaja selectiva, pueden llevar como pasajeros en las células cuando las células adquieren mutaciones/epimutations adicionales que proporcionan una ventaja proliferativa.
Etimología
El tumor del término se deriva de la Latina "tumere" a hincharse. Es similar a la Francés antiguo tumor (francés contemporáneo: Cancer). En la Commonwealth "tumor" de la ortografía se utiliza comúnmente, mientras que en Estados Unidos generalmente se escribe "tumor".
En su sentido médico tiene tradicionalmente una hinchazón anormal de la carne. El Enciclopedista médico romano Celso (ca 30 A.C. – 38 D.C.) describe los cuatro signos cardinales de aguda inflamación como tumor, dolor, calor, y rubor (inflamación, dolor, aumento de calor y enrojecimiento). Su tratado, De Medicina, se imprimió el primer libro médico en 1478 tras la invención de la imprenta de tipos móviles.
En inglés contemporáneo, el tumor de la palabra se utiliza a menudo como sinónimo para un crecimiento quístico (llenos de líquido) o neoplasma sólido (canceroso o no canceroso),[59] con otras formas de inflamación a menudo se refiere como inflamiento.[60]
Relacionada con los términos son comunes en la literatura médica, donde la tumefacción de los sustantivos y Tumescencia (derivado del adjetivo tumefied), son términos médicos actuales para la hinchazón no neoplásica. Este tipo de inflamación a menudo es causada por inflamación causada por trauma, infección y otros factores.
Los tumores pueden ser causados por condiciones distintas de una proliferación de células neoplásicas, sin embargo. Los quistes (por ejemplo, quistes sebáceos) son también contemplados como tumores, aunque no tienen células neoplásicas. Esto es estándar en terminología médica facturación (especialmente cuando la facturación para un crecimiento cuya patología tiene todavía ser determinado).
Véase también
- Antineoplaston
- Epigenética del cáncer
- Carcinogénesis
- Cáncer colorrectal
- Carcinoma
- Daños en el ADN (natural)
- Trastorno de deficiencias en la reparación de ADN
- Epigenética
- Silenciamiento del gen
- Inestabilidad genómica
- Oncología
- Evolución somática en cáncer
Referencias
- ^ Cooper GM (1992). Elementos de cáncer en humanos. Boston: Jones y Bartlett editores. p. 16. ISBN978-0-86720-191-8.
- ^ Taylor, Elizabeth J. (2000). Diccionario ilustrado de Dorland. (29 Ed.). Philadelphia: Saunders. p. 1184. ISBN0721662544.
- ^ Diccionario médico de Stedman (ed. 28 ed.). Philadelphia: Lippincott Williams & Wilkins. 2006. p. neoplasia. ISBN0781733901.
- ^ "Neoplams II". Organización Mundial de la salud. 19 de junio de 2014.
- ^ a b Abrams, Gerald. "Neoplasia". 23 de enero de 2012.
- ^ "Cáncer - actividad 1 - Glosario, página 4 de 5". Retrieved 2008-01-08.
- ^ "Neoplasias de células plasmáticas". WebMD. 16 de mayo de 2012. Retrieved 2008-01-08.
- ^ Willis, RA (1952). La propagación de los tumores en el cuerpo humano. London: Butterworth.[Página necesitado]
- ^ Lee ES, casillero J, Nalesnik M, Reyes J, Jaffe R, Alashari M, B Nour, Tzakis A, Dickman PS (enero de 1995). "La Asociación del virus Epstein - Barr con tumores del músculo liso que ocurren después del trasplante del órgano". N. Engl. J. Med. 332 (1): 19 – 25. Doi:10.1056/NEJM199501053320104. PMID7990861.
- ^ "Cáncer de páncreas: Glosario de términos". Retrieved 2008-01-08.
- ^ "Tumor". Dorland ilustrado diccionario médico (31 Ed.). Saunders. 2007. ISBN978-1-84972-348-0.
- ^ Ambrosi d., Mollica F (2002) "En la mecánica de un tumor en crecimiento". Revista Internacional de Ciencias de la ingeniería 40 (12): 1297 – 316. Doi:10.1016/S0020-7225 (02) 00014-9.
- ^ Volokh KY (septiembre de 2006). "Las tensiones en el cultivo de tejidos blandos". Acta Biomater 2 (5): 493 – 504. Doi:10.1016/j.actbio.2006.04.002. PMID16793355.
- ^ a b Bernstein C, Prasad AR Nfonsam V, Bernstei H (2013). "Daños en el ADN, la reparación del ADN y cáncer". Nuevas direcciones de investigación en la reparación del ADN. págs. 413 – 65. Doi:10.5772/53919. ISBN978-953-51-1114-6.
- ^ Bernstein, Carol (14 de enero de 2009). "Daños en el ADN y cáncer". SciTopics. 24 de septiembre de 2013.
- ^ Cunningham FH, Fiebelkorn S, Johnson M, Meredith C (noviembre de 2011). "Una novela aplicación del enfoque de margen de exposición: segregación de sustancias tóxicas del humo del tabaco". Food Chem Toxicol. 49 (11): 2921 – 33. Doi:10.1016/j.fct.2011.07.019. PMID21802474.
- ^ Kanavy él, Capaccioni MR (diciembre de 2011). La radiación ultravioleta y el melanoma. Semin Cutan Med Surg 30 (4): 222 – 8. Doi:10.1016/j.Sder.2011.08.003. PMID22123420.
- ^ Handa O, Y Naito, Yoshikawa T (2011). "Biología de redox y carcinogénesis gástrica: el papel del Helicobacter pylori". Redox Rep. 16 (1): 1 – 7. Doi:10.1179/174329211 X 12968219310756. PMID21605492.
- ^ Bernstein C, H Holubec, Bhattacharyya AK, H Nguyen, Payne CM, Zaitlin B, Bernstein H (agosto de 2011). "Carcinogenicidad de desoxicolato, un ácido biliar secundario". Arch Toxicol. 85 (8): 863 – 71. Doi:10.1007/s00204-011-0648-7. PMC3149672. PMID21267546.
- ^ Katsurano M, T Niwa, Yasui Y, Shigematsu Y, Yamashita S, H Takeshima, Lee MS, Kim YJ, Tanaka T, Ushijima T (enero de 2012). "La formación incipiente de un campo epigenético defecto en un modelo de colitis del ratón y no esenciales funciones de T - y B-células en la inducción de la metilación del ADN". Oncogén 31 (3): 342 – 51. Doi:10.1038/ONC.2011.241. PMID21685942.
- ^ Malkin d. (abril de 2011) "El síndrome de Li-fraumeni". Los genes cáncer 2 (4): 475 – 84. Doi:10.1177/1947601911413466. PMC3135649. PMID21779515.
- ^ Lichtenstein P, Holm NV, Verkasalo PK, Iliadou A, Kaprio J, Koskenvuo M, piniforme E, Skytthe A, Hemminki K (julio de 2000). "Factores ambientales y hereditarios en la causalidad de cáncer - análisis de cohortes de gemelos de Suecia, Dinamarca y Finlandia". N. Engl. J. Med. 343 (2): 78-85. Doi:10.1056/NEJM200007133430201. PMID10891514.
- ^ Halford S, Rowan A Sawyer E, Talbot I, Tomlinson (junio 2005). "O 6-metilguanina metiltransferasa en cáncer colorrectal: detección de las mutaciones, la pérdida de expresión y Asociación débil con G:C > A:T transiciones". Gut 54 (6): 797-802. Doi:10.1136/gut.2004.059535. PMC1774551. PMID15888787.
- ^ a b Shen L Kondo Y Rosner GL, Xiao L, Hernández NS, Vilaythong J, Houlihan PS, bajen RS, Prasad AR, Einspahr JG, Buckmeier J, Alberts DS, Hamilton SR, Issa JP (septiembre de 2005). "El campo y la metilación del promotor MGMT defecto en el cáncer colorrectal esporádico". J. nacional Cancer Inst. 97 (18): 1330 – 8. Doi:10.1093/jnci/dji275. PMID16174854.
- ^ Psofaki V, Kalogera C, Tzambouras N, D Stephanou, Tsianos E, Seferiadis K, Kolios G (julio de 2010). "Promotor metilación status of hMLH1, MGMT y CDKN2A/p16 en adenomas colorrectales". World j Gastroenterol. 16 (28): 3553 – 60. Doi:10.3748/wjg.V16.i28.3553. PMC2909555. PMID20653064.
- ^ a b c Lee KH, Lee JS, Nam JH, Choi C, Lee MC, Parque de CS, Juhng SW, Lee JH (octubre de 2011). "Estado de metilación del promotor de hMLH1 y hMSH2 genes MGMT en cáncer colorrectal asociado con secuencia adenoma-carcinoma." Langenbecks Arch Surg 396 (7): 1017 – 26. Doi:10.1007/s00423-011-0812-9. PMID21706233.
- ^ Entrada A, Sartore-Bianchi A, C Moutinho, Belotti A, Gonzalez K, Chirico G, Cassingena A, F Rusconi, Esposito A, Nichelatti M, Esteller M, Siena S (abril de 2013). "Promotor isla CpG hipermetilación de la enzima de reparación del ADN MGMT predice la respuesta clínica a la dacarbacina en una fase II estudio para el cáncer colorrectal metastásico". Clin. Cancer Res. 19 (8): 2265 – 72. Doi:10.1158/1078-0432.CCR-12-3518. PMID23422094.
- ^ Mokarram P, Zamani M, Kavousipour S, Naghibalhossaini F, Irajie C, Moradi Sarabi M, Hosseini SV (mayo de 2013). "Diferentes patrones de metilación del ADN de las dos distintas O6-metilguanina-DNA metiltransferasa (O6-MGMT) promotor regiones cáncer colorrectal". Análizar Biol Rep. 40 (5): 3851 – 7. Doi:10.1007/s11033-012-2465-3. PMID23271133.
- ^ Truninger K, Menigatti M, Luz J, Russell A, Haider R, Gebbers JO, Bannwart F, H Yurtsever, Neuweiler J, Riehle HM, MS Cattaruzza, Heinimann K, Schär P, Jiricny J, Marra G (mayo de 2005). "Análisis Immunohistochemical revela la alta frecuencia de PMS2 defectos en el cáncer colorrectal". Gastroenterología 128 (5): 1160 – 71. Doi:10.1053/j.gastro.2005.01.056. PMID15887099.
- ^ Valeri N, Gasparini P, Fabbri M, C Braconi, Veronese A, F Lovat, Adair B, Vannini Fanini F, Bottoni A, Costinean S, Sandhu SK, Nuovo GJ, aliso H, Gafa R, Calore F, Ferracin M, Lanza G, S Volinia, Negrini M, McIlhatton MA, D Amadori, R Fishel, Croce CM (abril de 2010). "Modulación de desajuste reparación y estabilidad genómica de miR-155". Proc. nacional Acad. Sci U.S.A. 107 (15): 6982 – 7. Doi:10.1073/pnas.1002472107. PMC2872463. PMID20351277.
- ^ a b c d e Facista A Nguyen H C Lewis, Prasad AR, Ramsey L, Zaitlin B, Nfonsam V, Jimmy RS, Bernstein H, Payne CM, Stern S, Oatman N, B Banerjee, Bernstein C (2012). "Expresión deficiente del DNA reparación enzimas en progresión temprana de cáncer de colon esporádicos". Genoma Integr 3 (1): 3. Doi:10.1186/2041-9414-3-3. PMC3351028. PMID22494821.
- ^ a b Narayanan L, Fritzell JA, Baker SM, Liskay RM, Glazer PM (abril de 1997). "Los niveles elevados de mutación en varios tejidos de ratones deficientes en el desajuste de ADN reparacion gene Pms2". Proc. nacional Acad. Sci U.S.A. 94 (7): 3122 – 7. Doi:10.1073/pnas.94.7.3122. PMC20332. PMID9096356.
- ^ a b HEGAN DC, Narayanan L, Jirik FR, Edelmann W, Liskay RM, Glazer PM (diciembre de 2006). "Diferentes patrones de inestabilidad genética en ratones deficientes en los genes de reparación de desajuste Pms2, Mlh1, Msh2, Msh3 y Msh6". Carcinogénesis 27 (12): 2402 – 8. Doi:10.1093/carcin/bgl079. PMC2612936. PMID16728433.
- ^ a b Tutt AN, van Oostrom CT, Ross GM, van H Steeg, A Ashworth (marzo de 2002). "Interrupción del Brca2 aumenta la tasa de mutación espontánea en vivo: sinergismo con radiación ionizante". EMBO Rep. 3 (3): 255 – 60. Doi:10.1093/EMBO-Reports/kvf037. PMC1084010. PMID11850397.
- ^ O ' hagan HM, Mohammad HP, Baylin SB (2008). "Roturas de doble hebra pueden iniciar silenciamiento génico y aparición de SIRT1 dependiente de metilación del ADN en una isla CpG promotor exógena". En heces, Jeannie T. PLoS Genet. 4 (8): e1000155. Doi:10.1371/Journal.pGEN.1000155. PMC2491723. PMID18704159.
- ^ Cuozzo C, Porcellini A, T Angrisano, Morano A, Lee B, Di Pardo A, S Messina, Iuliano R, Fusco A, Santillo señor, Muller MT, Chiariotti L, ME Gottesman, Avvedimento EV (julio de 2007). "Daños en el ADN, reparación dirigida por homología y la metilación del ADN". PLoS Genet. 3 (7): e110. Doi:10.1371/Journal.pGEN.0030110. PMC1913100. PMID17616978.
- ^ Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martínez P, Matthews N, Stewart A, P Tarpey, Varela I, B Phillimore, Begum S, McDonald ' s NQ, Butler A Jones D K Raine, Latimer C, Santos CR, Nohadani M, Eklund AC, B Spencer-Dene, Clark G, Pickering L, sello G, Gore M, Szallasi Z, J hacia abajo, Futreal PA, Swanton C (marzo de 2012). "Intratumor la heterogeneidad y la evolución ramificada revelaron ordenando multirregionalidad". N. Engl. J. Med. 366 (10): 883 – 92. Doi:10.1056/NEJMoa1113205. PMID22397650.
- ^ Matanza DP, Southwick HW, Smejkal W (septiembre de 1953). "Campo cancerization en epitelio escamoso estratificado oral; implicaciones clínicas de origen multicéntrico". Cáncer 6 (5): 963 – 8. Doi:10.1002/1097-0142 (195309) 6:5 < 963::AID-CNCR2820060515 > 3.0.CO;2-Q. PMID13094644.
- ^ Bernstein C, Bernstein H, Payne CM, Dvorak K, Garewal H (febrero de 2008). "Campo de defectos en la progresión de los cánceres del tracto gastrointestinal". Cáncer Lett. 260 (1 – 2): 1 – 10. Doi:10.1016/j.canlet.2007.11.027. PMC2744582. PMID18164807.
- ^ Nguyen H, C Loustaunau, Facista A, Ramsey L, Hassounah N, H Taylor, Jimmy R, Payne CM, Tsikitis VL, S Goldschmid, Banerjee B, Perini RF, Bernstein C (2010). "Deficiente Pms2, CcOI ERCC1, Ku86, en campo de defectos durante la progresión del cáncer de colon". J Exp de Vis (41): 1931. Doi:10.3791/1931. PMC3149991. PMID20689513.
- ^ Rubin H (marzo de 2011). "Y campo cancerization: los orígenes del cáncer preneoplásicas: campos hiperplásicas asintomáticos son precursores de la neoplasia y su progresión de los tumores puede ser rastreado por densidad de saturación en la cultura". Diseñó 33 (3): 224 – 31. Doi:10.1002/bies.201000067. PMID21254148.
- ^ Tsao JL, Yatabe Y, Salovaara R, Järvinen HJ, Mecklin JP, Aaltonen LA, Tavaré S, Shibata D (febrero de 2000). "Reconstrucción genética de las historias individuales del tumor colorrectal". Proc. nacional Acad. Sci U.S.A. 97 (3): 1236 – 41. Doi:10.1073/pnas.97.3.1236. PMC15581. PMID10655514.
- ^ a b c Vogelstein B, Papadopoulos N, VE Velculescu, Zhou S, Diaz LA, Kinzler KW (marzo de 2013). "Paisajes de genoma del cáncer". Ciencia 339 (6127): 1546 – 58. Doi:10.1126/science.1235122. PMC3749880. PMID23539594.
- ^ Lochhead P Chan en Nishihara R, Fuchs CS, Beck AH, Giovannucci E, efecto de campo Ogino S. Etiologic: revaloración del concepto de efecto de campo en progresión y predisposición al cáncer. Mod Pathol 2014 doi: 10.1038/modpathol.2014.81. [Epub ahead of print]
- ^ Svrcek M, Buhard O, Colas C, F Coulet, Dumont S, Massaoudi I, Lamri A Hamelin R Cosnes J, Oliveira C, Seruca R, Gaub MP, Legrain M, Collura A, O Lascols, inciso E, Fléjou JF, Duval (noviembre de 2010). "Tolerancia de metilación debido a un defecto del campo O6-metilguanina DNA metiltransferasa (MGMT) en la mucosa colónica: un paso iniciando en el desarrollo de los cánceres colorrectales de desajuste reparaciones deficientes". Gut 59 (11): 1516 – 26. Doi:10.1136/gut.2009.194787. PMID20947886.
- ^ Paluszczak J, P Misiak, Wierzbicka M, Woźniak A, Baer-Dubowska W (febrero de 2011). "Frecuente hipermetilación de DAPK, RARbeta, MGMT, RASSF1A y FHIT en carcinomas de células escamosas laríngeos y mucosa adyacente normal". Oral Oncol. 47 (2): 104 – 7. Doi:10.1016/j.oraloncology.2010.11.006. PMID21147548.
- ^ Zuo C, Zhang H, Spencer HJ, Vural E, Suen JY, Schichman SA, Smoller BR, MS Kokoska, Fan CY (octubre de 2009). Inestabilidad de microsatélites creciente y epigenética inactivación del gen hMLH1 en cabeza y cuello de células escamosas. Otolaryngol Head cuello Surg 141 (4): 484 – 90. Doi:10.1016/j.otohns.2009.07.007. PMID19786217.
- ^ Tawfik HM, El-Maqsoud NM, Hak BH, El-Sherbiny YM (2011). "Cabeza y cuello de células escamosas: desajuste reparación hipermetilación inmunohistoquímica y promotor del gen hMLH1". Soy J Otolaryngol 32 (6): 528-36. Doi:10.1016/j.amjoto.2010.11.005. PMID21353335.
- ^ Zou XP, Zhang B, Zhang XQ, Chen M, Cao J, Liu WJ (noviembre de 2009). "Hipermetilación del promotor de múltiples genes en lesiones precancerosas y adenocarcinoma gástrico temprana". Zumbido. Pathol. 40 (11): 1534 – 42. Doi:10.1016/j.humpath.2009.01.029. PMID19695681.
- ^ Wani M, Afroze D, Makhdoomi M, Hamid I, B Wani, Bhat G, R Wani, Wani K (2012). "Condición de promotor metilación de ADN reparar genes (hMLH1) en pacientes con carcinoma gástrico del Valle de Cachemira". Asia Pac. J. Cancer prev. 13 (8): 4177 – 81. Doi:10.7314/APJCP.2012.13.8.4177. PMID23098428.
- ^ Agarwal A, Polineni R, Hussein Z Vigoda Bhagat TD, S Bhattacharyya, A Maitra, Verma A (2012). "Papel de las alteraciones epigenéticas en la patogenesia del esófago de Barrett y adenocarcinoma del esófago". Int J Clin Exp Pathol 5 (5): 382 – 96. PMC3396065. PMID22808291.
- ^ B Hofstad, Vatn MH, Andersen SN, HUITFELD HS, Rognum T, S Larsen, Osnes M (septiembre de 1996). "Crecimiento de los pólipos colorrectales: redetection y evaluación de los pólipos durante un período de tres años". Gut 39 (3): 449 – 56. Doi:10.1136/gut.39.3.449. PMC1383355. PMID8949653.
- ^ Schmitt MW, Prindle MJ, Loeb LA (septiembre de 2012). "Implicaciones de la heterogeneidad genética en el cáncer". Ann N. Y. Acad. et al. 1267:: 110 – 6. Doi:10.1111/j.1749-6632.2012.06590.x. PMC3674777. PMID22954224.
- ^ Lander ES, Linton LM, Birren B, C Nusbaum, Zody MC, J Baldwin, Devon K, Dewar K, Doyle M, FitzHugh W, et al.. (Febrero de 2001). "La secuencia inicial y análisis del genoma humano". Naturaleza 409 (6822): 860 – 921. Doi:10.1038/35057062. PMID11237011.
- ^ Yost SE Smith EN Schwab RB, Bao L, Jung H, Wang X, E de Voest, Pierce JP, Messer K, Parker BA, O Harismendy, Frazer KA (agosto de 2012). "Identificación de las mutaciones somáticas alta confianza en la secuencia del genoma entero de especímenes de cáncer de pecho con formol". Ácidos nucleic Res. 40 (14): e107. Doi:10.1093/nar/gks299. PMC3413110. PMID22492626.
- ^ Berger MF, Hodis E, TP Heffernan, Deribe YL, Lawrence MS, Protopopov A, E Ivanova, Watson IR, Nickerson E, Ghosh P, Zhang H, Zeid R, Ren X, Cibulskis K, Sivachenko AY, Wagle N, lechón A, Sougnez C, Onofrio R, Ambrogio L, Auclair D, T Fennell, Carter SL, Y secador, Stojanov P, cantante MA, Voet D, Jing R, Saksena G, Barretina J, Ramos AHPugh TJ, N Stransky, Parkin M, Winckler W, Mahan S, Ardlie K, J Baldwin, Wargo J, Schadendorf D, Meyerson M, Gabriel SB, Golub TR, Wagner SN, Lander ES, Getz G, barbilla L, LA Garraway (mayo de 2012). "Melanoma genoma secuencia revela frecuentes mutaciones PREX2". Naturaleza 485 (7399): 502 – 6. Doi:10.1038/nature11071. PMC3367798. PMID22622578.
- ^ Roach JC, G Glusman, Smit AF, et al. (Abril de 2010). "Análisis de la herencia genética en un cuarteto familiar por la secuencia del genoma entero". Ciencia 328 (5978): 636-9. Doi:10.1126/science.1186802. PMC3037280. PMID20220176.
- ^ Campbell CD, Chong JX, Malig M, et al. (Noviembre de 2012). "Estimación de la tasa de mutación humana usando autozygosity en una población fundadora". NAT Genet. 44 (11): 1277 – 81. Doi:10.1038/ng.2418. PMC3483378. PMID23001126.
- ^ Tumor en Enciclopedia médica
- ^ "Hinchazón". MedlinePlus enciclopedia médica. 14 de octubre de 2012.
|
||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||