Fibrosis quística
| Fibrosis quística | |
|---|---|
| Clasificación y recursos externos | |

Dedos hipocráticos en los dedos de una persona con fibrosis quística
|
|
| CIE-10 | E84in |
| CIE-9 | 277.0 |
| OMIM | 219700 |
| DiseasesDB | 3347 |
| MedlinePlus | 000107 |
| eMedicine | PED/535 |
| Paciente UK | Fibrosis quística |
| Malla | D003550 |
Fibrosis quística (CF), también conocido como mucovisidosis, es una autosómico recesivo ligado al sexo trastorno genético que afecta sobre todo el pulmonesy también el páncreas, hígado, y intestino. Dificultad para respirar es el síntoma más grave y da como resultado de frecuentes infecciones pulmonares. Otros síntomas— incluyendo infecciones sinusales, pobre crecimiento, y infertilidad— afectan a otras partes del cuerpo.
CF es causada por una de muchas mutaciones diferentes en el Gene[1] para el proteína regulador de la conductancia transmembrana de la fibrosis quística (CFTR). Esta proteína es necesaria para regular los componentes de sudor, digestivo fluidos, y moco. Personas sanas tienen dos copias del trabajo del gen CFTR. Los portadores tienen una copia de trabajo. Las personas con FQ no tienen ninguna copia de trabajo. Por lo tanto tiene CF autosómico recesivo ligado al sexo herencia. El mecanismo subyacente es transporte anormal de cloruro de y sodio a través de la epitelio, que es la capa celular que cubre las membranas de los órganos. Esto conduce a las secreciones gruesas y viscosas.[2] Las personas con fibrosis quística se pueden diagnosticar antes del nacimiento por las pruebas genéticas, o por un prueba del sudor[3] en la primera infancia.
Las infecciones pulmonares son tratadas con antibióticos y otros medicamentos. En última instancia, trasplante de pulmón a menudo es necesario como CF empeora. La esperanza media de vida es de 37 a 40 años en Estados Unidos.[4] CF es más común entre personas de Central y Europea del norte ascendencia, pero ocurre en muchos grupos diferentes alrededor del mundo. Es más rara entre Asiáticos y el Puentes media.[5][6][7][8]
El nombre fibrosis quística se refiere a la característica marcar con una cicatriz (fibrosis) y quiste la formación dentro de la páncreasprimero reconocida en la década de 1930.[9]
Contenido
- 1 Signos y síntomas
- 1.1 Los pulmones y los senos paranasales
- 1.2 Gastrointestinal
- 1.3 Sistema endocrino
- 1.4 Infertilidad
- 2 Causa
- 3 Fisiopatología
- 3.1 Infecciones crónicas
- 4 Diagnóstico y seguimiento
- 4.1 Prenatal
- 5 Gestión
- 5.1 Antibióticos
- 5.2 Otros tratamientos para la enfermedad pulmonar
- 5.3 Trasplante
- 5.4 Otros aspectos
- 6 Pronóstico
- 6.1 Calidad de vida
- 7 Epidemiología
- 7.1 Evolución
- 8 Historia
- 9 Investigación
- 9.1 Terapia génica
- 9.2 Pequeñas moléculas
- 10 Sociedad y cultura
- 11 Referencias
- 12 Lectura adicional
- 13 Enlaces externos
Signos y síntomas
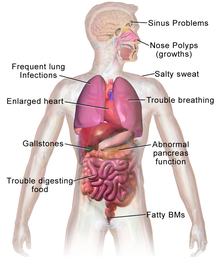
Los principales signos y síntomas de la fibrosis quística son sabor salado piel,[10] pobre crecimiento y aumento de peso pobre a pesar de la toma de comida normal,[11] acumulación de moco espeso y pegajoso,[12] infecciones respiratorias frecuentes y tos o dificultad para respirar.[13] Los machos pueden ser infértil debido a ausencia congénita de conductos deferentes.[14] Los síntomas aparecen a menudo en infancia y niñez, tales como obstrucción intestinal debido a íleo meconial en los recién nacidos.[15] A medida que los niños crecen, debe ejercer para liberar el moco en los alvéolos.[16] Ciliado células epiteliales en el paciente tiene una proteína mutada que conduce a la producción de moco anormalmente viscosa.[12] El pobre crecimiento en los niños típicamente se presenta como una incapacidad para ganar peso o altura al mismo ritmo que sus compañeros y en ocasiones no se diagnostica hasta que se inicie la investigación para un crecimiento pobre. Las causas de la falta del crecimiento son multifactoriales e incluyen infección pulmonar crónica, mala absorción de nutrientes a través del tracto gastrointestinal y aumentaron de la demanda metabólica debido a una enfermedad crónica.[11]
En casos raros, la fibrosis quística puede manifestarse como un trastorno de la coagulación. La vitamina K es absorbida normalmente de la leche materna, fórmula y alimentos sólidos después. Esta absorción está deteriorada en algunos pacientes con fibrosis quística. Los niños pequeños son especialmente sensibles a vitamina K trastornos de malabsorción porque sólo una muy pequeña cantidad de vitamina K cruza la placenta, dejando al niño con reservas muy bajas y una capacidad limitada para absorber vitamina K de fuentes dietéticas después del nacimiento. Porque los factores II, VII, IX y X (factores de coagulación) – dependiente de la vitamina K, bajos niveles de vitamina K pueden resultar en problemas de coagulación. En consecuencia, cuando un niño presenta hematomas inexplicables, una evaluación de la coagulación puede estar justificada para determinar si existe una enfermedad subyacente.[17]
Los pulmones y los senos paranasales

Verde = Pseudomonas aeruginosa
Marrón = Estafilococo áureo
Azul = Haemophilus influenzae
Rojo = Burkholderia cepacia complejo
Enfermedad pulmonar resulta de la obstrucción de las vías respiratorias debido a la acumulación de moco, disminuido mucociliary como resultado inflamación.[18][19] Inflamación e infección causan cambios estructurales y lesiones en los pulmones, llevando a una variedad de síntomas. En las primeras etapas, tos incesante, abundante flema producción y disminución de la capacidad de ejercer son comunes. Muchos de estos síntomas ocurren cuando bacterias que normalmente habitan el moco espeso crecen fuera de control y causar neumonía. En etapas posteriores, los cambios en la arquitectura del pulmón, tales como la patología en las vías aéreas principales (bronquiectasia), exacerbar aún más las dificultades en la respiración. Otros síntomas incluyen tos con sangre)hemoptisis), alta presión arterial en el pulmón (hipertensión pulmonar), insuficiencia cardíaca, las dificultades que consiguen suficiente oxígeno para el cuerpo (hipoxia) e insuficiencia respiratoria que requiere ayuda con la respiración tales como máscaras, presión positiva binivel vía aérea máquinas o ventiladores.[20] Estafilococo áureo, Haemophilus influenzae, y Pseudomonas aeruginosa son los tres organismos más comunes que causan infecciones pulmonares en pacientes con FQ.[19] Además de las infecciones bacterianas típicas, las personas con FQ más comúnmente desarrollan otros tipos de enfermedad pulmonar. Entre ellos se encuentra aspergilosis broncopulmonar alérgica, en el cual la respuesta del cuerpo a la común hongo Aspergillus fumigatus causas empeoramiento de problemas respiratorios. Otra es la infección con Mycobacterium avium complejo (MAC), un grupo de bacterias relacionadas con tuberculosis, que puede causar mucho daño a los pulmones y no responde a los antibióticos comunes.[21]
Moco en la senos paranasales es igualmente gruesa y también pueden causar la obstrucción de los conductos del seno, conduce a la infección. Esto puede causar dolor facial, fiebre, secreción nasal, y dolores de cabeza. Las personas con FQ pueden desarrollar el crecimiento excesivo de los tejidos nasales (pólipos nasales) debido a la inflamación de las infecciones sinusales crónicas.[22] Los pólipos sinonasal recurrente pueden ocurrir en tanto como de 10% a 25% de los pacientes con FQ.[19] Estos pólipos pueden bloquear los conductos nasales y aumentar las dificultades respiratorias.[23][24]
Las complicaciones cardiorespiratorias son la causa más común de muerte (~ 80%) en pacientes en la mayoría de los centros CF en los Estados Unidos.[19]
Gastrointestinal
Antes de prenatal y evaluación del recién nacido, la fibrosis quística fue diagnosticada a menudo cuando un recién nacido no lograron pasar las heces (meconio). Meconio puede bloquear completamente el intestinos y causar una enfermedad seria. Esta condición, llamada íleo meconial, se produce en 5 – 10%[19][25] de los recién nacidos con CF. en adición, la saliente del interno rectal membranas)prolapso rectal) es más común, que ocurre en hasta 10% de los niños con fibrosis quística,[19] y es causada por el aumento del volumen fecal, desnutrición, y aumento de la presión intra – abdominal debido a la tos.[26]
La mucosidad espesa en los pulmones tiene una contraparte en las secreciones densas de los páncreas, un órgano responsable de proporcionar jugos digestivos ayudan a descomponer el alimento. Estas secreciones bloquean el exocrina movimiento de las enzimas digestivas en el duodeno y causar daños irreversibles en el páncreas, a menudo con inflamación dolorosa (pancreatitis).[27] El conductos pancreáticos son totalmente enchufado en casos más avanzados, que generalmente se observa en más viejos niños o adolescentes.[19] Esto causa la atrofia de las glándulas exocrinas y fibrosis progresiva.[19]
La falta de enzimas digestivas provoca dificultad para absorber los nutrientes con su posterior excreción en las heces, un trastorno conocido como malabsorción. Malabsorción conduce a desnutrición pobre crecimiento y desarrollo debido a la pérdida de calorías. Resultante hipoproteinemia puede ser lo suficientemente graves como para causar edema generalizado.[19] Las personas con fibrosis quística tienen también dificultades absorber las vitaminas liposolubles A, D, E, y K.
Además de los problemas de páncreas, más experimentan de pacientes con fibrosis quística ardor de estómago, obstrucción intestinal por invaginación intestinal, y estreñimiento.[28] Las personas mayores con FQ pueden desarrollar síndrome de obstrucción intestinal distal Cuando las heces espesadas causan obstrucción intestinal.[29]
Insuficiencia Pancreática Exocrina se produce en la mayoría (85% a 90%) de los pacientes con FQ.[19] Se asocia principalmente con mutaciones de CFTR "severas", donde ambos alelos son absolutamente frizados (por ejemplo ΔF508/ΔF508).[19] Ocurre en 10% a 15% de los pacientes con una "severa" y una "leve" mutación de CFTR donde todavía hay una pequeña actividad CFTR, o donde existen dos mutaciones de CFTR "suaves".[19] En estos casos más leves, hay todavía suficiente función exocrina pancreática así que no es necesaria la suplementación con enzima.[19] Generalmente no hay otros GI complicaciones en fenotipos páncreas-suficiente, y en general, estas personas suelen tienen excelente crecimiento y desarrollo.[19] A pesar de ello, idiopática pancreatitis crónica puede ocurrir en un subconjunto de páncreas-suficientes personas con fibrosis quística y se asocia con dolor abdominal recurrente y complicaciones potencialmente mortales.[19]
Las secreciones densas también pueden causar problemas hepáticos en los pacientes con FQ. Bilis secretada por el hígado para ayudar en la digestión puede bloquear el conductos biliares, conduce a daños en el hígado. Con el tiempo, esto puede conducir a la cicatrización y nodularidad (cirrosis). El hígado es incapaz de eliminar la sangre de toxinas y no hace importante proteínas, como los responsables de la coagulación de la sangre.[30][31] Enfermedad del hígado es la tercera más frecuente causa de muerte asociada con fibrosis quística.[19]
Sistema endocrino
El páncreas contiene el islotes de Langerhans, que son responsables de la insulina, una hormona que ayuda a regular la sangre glucosa. Daño del páncreas puede conducir a la pérdida del islote células, llevando a un tipo de diabetes que es única para aquellos que padecen la enfermedad.[32] Esto diabetes relacionada con la fibrosis quística (CFRD) comparte características que pueden encontrarse en tipo 1 y tipo 2 los diabéticos, y es una de las principales complicaciones nonpulmonary de la FQ.[33] La vitamina D está implicada en calcio y fosfato regulación. Mala absorción de la vitamina D de la dieta debido a la mala absorción puede conducir a la enfermedad de los huesos osteoporosis en la que los huesos debilitados son más susceptibles a fracturas.[34] Además, las personas con FQ a menudo desarrollan dedos hipocráticos de sus dedos y dedos de los pies debido a los efectos de una enfermedad crónica y bajo nivel de oxígeno en sus tejidos.[35][36]
Infertilidad
Infertilidad afecta a hombres y mujeres. Al menos 97% de los hombres con fibrosis quística son estériles, pero no estéril y pueden tener hijos con técnicas de reproducción asistida.[37] Es la principal causa de infertilidad en los hombres con fibrosis quística ausencia congénita de conductos deferentes (que normalmente se conecta el testículos para el Conductos Eyaculadores de la pene), pero potencialmente también por otros mecanismos como la causa azoospermia, Teratozoospermia, y oligoasthenospermia.[38] Muchos hombres encontrados que la ausencia congénita de conductos deferentes durante la evaluación de la infertilidad tienen una forma suave, no diagnosticada previamente de la FQ.[39] Aproximadamente el 20% de las mujeres con fibrosis quística tienen dificultades de fertilidad debido al engrosamiento moco cervical o desnutrición. En casos severos, la desnutrición afecta ovulación y causas amenorrea.[40]
Causa

CF es causada por una mutación en el Gene regulador de la conductancia transmembrana de la fibrosis quística (CFTR). La mutación más común, ΔF508, es una eliminación (Δ lo que significa eliminación) de tres nucleótidos[41] Eso resulta en una pérdida del aminoácido fenilalanina (F) en la posición 508a en la proteína. Esta mutación representa las dos terceras partes (66-70%[19]) de CF casos en todo el mundo y el 90% de casos en el Estados Unidos; Sin embargo, hay más de 1500 otras mutaciones que pueden producir CF.[42] Aunque la mayoría de las personas tiene dos copias del trabajo (alelos) del gen CFTR, sólo es necesaria para evitar la fibrosis quística. CF se desarrolla cuando ninguno de los dos alelos pueden producir una proteína CFTR funcional. Por lo tanto, CF es considerado un enfermedad autosómica recesiva.
El Gen CFTR, en el q31.2 lugar geométrico de cromosoma 7, es 230.000 pares de bases de largo y crea una proteína que es 1.480 los aminoácidos largo. Sub más específicamente la ubicación es entre pares 117,120,016 a 117,308,718 en el brazo largo del cromosoma 7, región 3, grupo 1, grupo 2, representado como 7q31.2. Estructuralmente, la CFTR es un tipo de gen conocido como un Gene ABC.[20] El producto de este gen (el CFTR) es un canal de iones cloruro importante en la creación de sudor, digestivo jugos y moco. Esta proteína posee dos ATP-hidrolización Dominios, que permite que la proteína a utilizar energía en forma de ATP. También contiene dos dominios que comprende 6 Alfa hélices cada uno, que permiten la proteína cruzar la membrana celular. Un regulador sitio de Unión en la proteína permite la activación por fosforilación, principalmente por quinasa dependiente de cAMP.[20] El carboxilo terminal de la proteína está anclada a la citoesqueleto por un PDZ interacción de dominio.[43]
Además, hay evidencia creciente de que los modificadores genéticos además de CFTR modulan la frecuencia y la severidad de la enfermedad. Un ejemplo es lectina de unión a Manano, que está implicado en inmunidad innata facilitando fagocitosis de los microorganismos. Polimorfismos en uno o ambos Manano-lectina alelos que resultan en bajos niveles circulantes de la proteína se asocian con un triple y mayor riesgo de enfermedad pulmonar en etapa terminal, así como una carga creciente de infecciones bacterianas crónicas.[19]
Fisiopatología
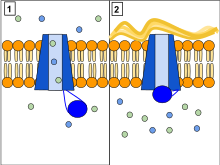
Existen varias mutaciones en el CFTR gen y diversas mutaciones causan defectos diferentes en la proteína CFTR, a veces causando una enfermedad más leve o más severa. Estos defectos de proteína son también objetivos para las drogas que a veces pueden restaurar su función. ΔF508-CFTR, que ocurre en > 90% de los pacientes en los Estados Unidos, crea una proteína que no doblez normalmente y se degrada por la célula. Resultan de otras mutaciones en proteínas que son demasiado cortas (truncado) porque producción se terminó prematuramente. Otras mutaciones producen proteínas que: No use energía normalmente, no permiten cloruro de, Yoduro, y tiocianato para cruzar la membrana apropiadamente,[44] degradar a un ritmo más rápido de lo normal. Las mutaciones también pueden llevar a menos copias de la proteína CFTR se producen.[20]
La proteína creada por este gen está anclada a la membrana externa de células En glándulas sudoríparas, pulmones, páncreas y todas otras glándulas exocrinas restantes en el cuerpo. La proteína atraviesa esta membrana y actúa como un canal conexión de la parte interior de la célula (citoplasma) para el líquido circundante. Este canal es principalmente responsable de controlar el movimiento de halógenos de dentro a fuera de la célula; Sin embargo, en los conductos de sudor facilita el movimiento del cloruro del sudor en el citoplasma. Cuando la proteína CFTR no funciona, cloruro y tiocianato[45] están atrapados dentro de las células en las vías respiratorias y del exterior en la piel. Entonces hypothiocyaniteOSCN, no puede ser producida por el sistema de defensa inmune.[46][47] Porque es cloruro cargado negativamente, esto crea una diferencia de potencial eléctrico dentro y fuera de la célula causando cationes para cruzar a la célula. El sodio es el catión más comunes en el espacio extracelular y la combinación de sodio y cloruro, crea el Sal, que se pierde en grandes cantidades en el sudor de las personas con CF. esto perdió sal forma la base para la prueba de sudor.[20]
La mayoría de los daños en la FQ es debido al bloqueo de los estrechos pasajes de órganos afectados con secreciones espesadas. Estos bloqueos llevan a remodelación y acumulado de infección en el pulmón, daño por enzimas digestivas en el páncreas, la obstrucción de los intestinos por las heces gruesas, etc.. Existen varias teorías sobre cómo los defectos en la función celular y de proteínas causan los efectos clínicos. Una teoría es que la falta de halógeno y Pseudohalógeno (principalmente, cloruro, yoduro y tiocianato) saliendo a través de la proteína CFTR conduce a la acumulación de moco en los pulmones más viscoso, rico en nutrientes que permite que las bacterias que esconderse del cuerpo sistema inmune. Otra teoría es que la falta de proteína CFTR conduce a un aumento paradójico en la absorción de sodio y cloruro, que, conduciendo a la reabsorción de agua creciente, crea moco deshidratado y grueso. Otra teoría más es ese movimiento anormal cloruro hacia fuera de la célula conduce a la deshidratación de las secreciones pancreáticas, secreciones biliares, moco, etc..[20]
Infecciones crónicas
Los pulmones de las personas con fibrosis quística son colonizados e infectados por bacterias desde muy temprana edad. Estas bacterias, que a menudo extensión entre las personas con FQ, prosperan en la mucosa alterada, que se acumula en las vías respiratorias pequeñas de los pulmones. Esta mucosidad conduce a la formación de microambientes bacterianas conocida como biofilms son difíciles para las células inmunitarias y antibióticos para penetrar. Secreciones viscosas y las infecciones respiratorias persistentes repetidamente dañan el pulmón por remodelación gradualmente las vías respiratorias, que hace aún más difícil de erradicar la infección.[48]
Con el tiempo, tanto los tipos de bacterias y sus características individuales cambian en individuos con CF. en la etapa inicial, las bacterias comunes tales como Estafilococo áureo y Haemophilus influenzae colonizar e infectar los pulmones.[19] Con el tiempo, Pseudomonas aeruginosa (y a veces Burkholderia cepacia) domina. Por 18 años de edad, albergan el 80% de los pacientes con FQ clásico P. aeruginosay el puerto de 3.5% B. cepacia.[19] Una vez dentro de los pulmones, estas bacterias adaptan al medio ambiente y desarrollar resistencia comúnmente utilizado antibióticos. Pseudomonas pueden desarrollar características especiales que permiten la formación de grandes colonias, conocido como "mucoide" Pseudomonas, que raramente se ven en las personas que no tienen CF.[48]
Una forma de infección se separa es por pasar entre diferentes personas con fibrosis quística.[49] En el pasado, las personas con FQ a menudo participaron en campamentos de verano"CF" y otras reuniones recreativas.[50][51] Hospitales agrupan (tales como los pacientes con FQ en zonas comunes y equipo rutina nebulizadores)[52] No se esterilizó entre pacientes individuales.[53] Esto condujo a la transmisión de cepas más peligrosas de bacterias entre grupos de pacientes. Como resultado, las personas con FQ son rutinariamente aisladas unos de otros en el ámbito profesional de la salud y los prestadores de servicios se les animados a usar guantes y batas al examinar a los pacientes con FQ de limitar la propagación de cepas bacterianas virulentas.[54]
Los pacientes con FQ pueden tener también sus vías respiratorias crónicamente colonizadas por hongos filamentosos (tales como Aspergillus fumigatus, Scedosporium apiospermum, Aspergillus terreus) o levaduras (tales como Candida albicans); otros hongos filamentosos aislados menos comúnmente incluyen Aspergillus flavus y Aspergillus nidulans (ocurrir transitoriamente en las secreciones respiratorias CF), y Exophiala dermatitidis y Scedosporium prolificans (crónica de las vías respiratorias-colonizadores); algunos hongos filamentosos como Penicillium emersonii y Acrophialophora Polyporales se encuentran en pacientes casi exclusivamente en el contexto de la FQ.[55] Mucociliar defectuoso separación recordarmos CF se asocia con trastornos inmunológicos locales. Además, el tratamiento prolongado con antibióticos y el uso de tratamientos con corticosteroides puede también facilitar crecimiento fúngico. Aunque la relevancia clínica de la colonización de hongos en las vías respiratorias sigue siendo un tema de debate, hongos filamentosos pueden contribuir a la respuesta inflamatoria local y por lo tanto para el progresivo deterioro de la función pulmonar, como sucede a menudo con aspergilosis broncopulmonar alérgica (ABPA) – la enfermedad fúngica más común en el contexto de la FQ, que implica una respuesta inmune Th2 impulsada por Aspergillus.[55][56]
Diagnóstico y seguimiento

La fibrosis quística puede ser diagnosticada por muchos diferentes métodos incluyendo evaluación del recién nacido, prueba del sudor, y las pruebas genéticas.[3] A partir de 2006 en Estados Unidos, 10 por ciento de los casos se diagnostican poco después del nacimiento como parte de programas de cribado de recién nacido. La pantalla recién inicialmente medidas de concentración de sangre elevado tripsinógeno inmunorreactivo.[57] Nacidos con una pantalla de recién nacido anormal necesitan una prueba de sudor para confirmar el diagnóstico de CF. En muchos casos, un padre hace el diagnóstico porque el niño sabe salado.[19] Tripsinógeno pueden aumentar los niveles en individuos que tienen una sola copia mutada la CFTR gen (portadores) o, en casos raros, en individuos con dos copias normales de la CFTR Gene. Debido a estos falsos positivosProyección CF en los recién nacidos puede ser polémico.[58][59] Mayoría de Estados y países no pantalla para CF rutinariamente en el nacimiento. Por lo tanto, la mayoría de las personas son diagnosticadas después de síntomas (por ejemplo enfermedad sinopulmonary y manifestaciones GI[19]) solicitar una evaluación para la fibrosis quística. La forma más comúnmente usada de pruebas es la prueba de sudor. Prueba de sudor implica la aplicación de un medicamento que estimula la sudoración)pilocarpina). Para entregar el medicamento a través de la piel, iontoforesis se utiliza, por el que uno electrodo se coloca sobre el medicamento aplicado y un corriente eléctrica se pasa un electrodo separado en la piel. El sudor resultante es entonces recogido en papel de filtro o en un tubo capilar y analizado por cantidades anormales de sodio y cloruro de. Las personas con FQ han aumentado cantidades de sodio y cloruro en el sudor. En cambio, las personas con FQ tienen menos tiocianato y hypothiocyanite en su saliva[60] y el moco (Banfi et al.). CF también puede ser diagnosticado por la identificación de mutaciones en el gen CFTR.[61]
Las personas con FQ pueden figurar en un registro de enfermedades que permite a los investigadores y médicos para seguimiento de resultados en salud e identificar a candidatos para ensayos clínicos.[62]
Prenatal
Las parejas que están embarazadas o planeando que un embarazo puede han probado para que las mutaciones del gene CFTR determinar el riesgo de que su hijo nacerá con fibrosis quística. La prueba se realiza típicamente primero en uno o ambos progenitores y, si es alto el riesgo de la FQ, prueba en la feto se lleva a cabo. El Colegio Americano de Obstetras y ginecólogos (ACOG) recomienda la prueba para las parejas que tienen un personal o cerca de antecedentes familiares de CF, y recomiendan eso prueba del portador se ofreció a todas las parejas caucásico y ponerse a disposición de las parejas de otros orígenes étnicos.[63]
Por el desarrollo de la FQ en el feto requiere cada padre a pasar una copia mutada del gen CFTR y porque pruebas CF es caro, la prueba se realiza con frecuencia inicialmente en uno de los padres. Si las pruebas muestran que padre es un portador de mutación de gen CFTR, el otro padre está testado calcular el riesgo de que sus hijos tengan CF. CF puede resultar de más de 1 mil mutaciones diferentes, y a partir de 2006 no es posible poner a prueba para cada uno. Prueba analiza la sangre para las mutaciones más comunes como ΔF508 — pruebas disponibles comercialmente la mayoría buscan 32 o menos mutaciones diferentes. Si una familia tiene una mutación común conocida, se pueden realizar proyecciones especificas para esa mutación. Porque no todas las mutaciones conocidas se encuentran en las pruebas actuales, una pantalla negativa no garantiza que un niño no tenga CF.[64]
Durante el embarazo, la prueba puede realizarse en el placenta (muestreo de vellosidades coriónicas) o el líquido que rodea al feto (amniocentesis). Sin embargo, muestreo de vellosidades coriónicas tiene un riesgo de muerte fetal de 1 en 100 y amniocentesis de 1 de cada 200;[65] un estudio reciente ha indicado que esto puede ser mucho menor, aproximadamente 1 en 1.600.[66]
Económicamente, para parejas de portador de fibrosis quística, comparando el diagnóstico genético preimplantacional (DGP) con la concepción natural (NC) seguido por la prueba prenatal y el aborto de embarazos afectados, PGD proporciona beneficios económicos netos hasta una edad materna de aproximadamente 40 años, después de que Carolina del norte, la prueba prenatal y el aborto tiene mayor beneficio económico.[67]
Gestión
Aunque no hay ninguna cura para la fibrosis quística, existen varios métodos de tratamiento. El tratamiento de la fibrosis quística ha mejorado significativamente en los últimos 70 años. Mientras que los recién nacidos con fibrosis quística hace 70 años hubiera sido improbables vivir más allá de su primer año, infantes tienen probabilidades de vivir hasta la edad adulta. Los recientes avances en el tratamiento de la fibrosis quística han significado que un individuo con fibrosis quística puede vivir una vida más plena que menos gravado por su condición. Los pilares de la gestión son tratamiento proactiva de infección de las vías respiratoriasy el fomento de la buena nutrición y un estilo de vida activo. Rehabilitación pulmonar como una gestión de la fibrosis quística continúa a lo largo de la vida del paciente y tiene como objetivo maximizar la función del órgano y por lo tanto la calidad de vida. A lo mejor, los tratamientos actuales retrasan la disminución de la función del órgano. Debido a la amplia variación de síntomas de la enfermedad, el tratamiento típicamente ocurre en los centros multidisciplinarios de especialista y se adapta al individuo. Objetivos para la terapia de la pulmones, tracto gastrointestinal (incluyendo suplementos de enzimas pancreáticas), la órganos reproductivos (incluyendo tecnología de reproducción asistida (ARTE)) y apoyo psicológico.[57]
El aspecto más consistente de la terapia en la fibrosis quística es limitar y tratar el daño pulmonar causado por la mucosidad espesa y la infección, con el objetivo de mantener calidad de vida. Intravenoso, inhalado, y los antibióticos orales se utilizan para tratar las infecciones crónicas y agudas. Dispositivos mecánicos y medicamentos para inhalación se utilizan para modificar y borrar el moco espesado. Estas terapias, al mismo tiempo eficaz, pueden ser sumamente lentos para el paciente. Una de las batallas más importantes que enfrentan los pacientes con FQ es encontrar el tiempo para cumplir con los tratamientos prescritos al mismo tiempo equilibrar una vida normal.
Además, las terapias tales como trasplante y terapia génica objetivo es curar algunos de los efectos de la fibrosis quística. La terapia génica pretende introducir CFTR normal a las vías respiratorias. Teóricamente, este proceso debe ser simple como las vías respiratorias es fácilmente accesible y hay sólo un defecto genético único para corregir. Existen dos mecanismos de introducción de gen CFTR involucrado, el primer uso de un vector viral (adenovirus, virus adeno-asociado o retro virus) y en segundo lugar el uso de liposoma. Sin embargo, existen algunos problemas asociados con estos métodos que implican eficacia (de liposomas escaso contenido en proteínas) y entrega (virus provoca una respuesta inmunitaria).
Antibióticos
Muchos pacientes con FQ están en uno o más antibióticos en todo momento, incluso cuando está saludable, profiláctico suprimir la infección. Los antibióticos son absolutamente necesarios cuando se sospecha de neumonía o ha habido una sensible disminución en la función pulmonar, y es generalmente elegido basado en los resultados de un análisis de esputo y respuesta pasados del paciente. Esta terapia prolongada con frecuencia requiere hospitalización y la inserción de una permanente más IV tales como un catéter central insertado periféricamente (La línea PICC) o Port-a-Cath. Terapia inhalada con antibióticos tales como Tobramicina, Colistina, y aztreonam a menudo se administra durante meses a la vez para mejorar la función pulmonar impidiendo el crecimiento de bacterias colonizadas.[68][69][70] Tratamiento antibiótico inhalado ayuda a la función pulmonar mediante la lucha contra la infección, pero también tiene desventajas significativas como el desarrollo de resistencia a los antibióticos, tinnitus y cambios en la voz.[71] Los antibióticos orales como la ciprofloxacina o azitromicina se administra para ayudar a prevenir la infección o para controlar la infección en curso.[72] El aminoglucósido los antibióticos (e.g. tobramycin) pueden causar pérdida de la audición, daño a la sistema de equilibrio En oído interno o problemas renales con el uso a largo plazo.[73] Para evitar estos efectos secundarios, la cantidad de antibióticos en la sangre son rutinariamente medido y ajustado en consecuencia.
Otros tratamientos para la enfermedad pulmonar
Se utilizan diversas técnicas mecánicas para desalojar esputo y favorecer la expectoración. En el ámbito hospitalario, fisioterapia torácica (CPT) se utiliza; un terapeuta respiratorio percusses pecho de un individuo con sus manos varias veces al día, para aflojar las secreciones. Los dispositivos que recrean esta terapia percusiva incluyen el Chaleco ThAIRapy y el ventilador percusiva intrapulmonar (IPV). Métodos más nuevos tales como Ventilación bifásico coraza, y modo de separación asociados disponible en estos dispositivos, integrar una fase de asistencia para la tos, así como una fase de vibración para desalojar las secreciones. Estos son portátiles y adaptado para el uso casero.[74]
Aerosol de los medicamentos que ayudan a aflojar las secreciones incluyen dornasa alfa y soluciones hipertónicas solución salina.[75] Dornasa es un recombinante humano Desoxirribonucleasa, que descompone el ADN en el esputo, disminuyendo su viscosidad.[76] Denufosol es un fármaco en fase de investigación que se abre un canal alternativo cloruro, ayudando a licuar el moco.[77]
Como pulmón enfermedad empeora, asistencia respiratoria mecánica puede ser necesario. Las personas con FQ pueden necesitar usar máscaras especiales por la noche que ayudan a empujar aire en sus pulmones. Estas máquinas, conocidas como presión positiva binivel vía aérea Ventiladores (BiPAP), ayudan a prevenir los niveles de oxígeno bajo en la sangre durante el sueño. BiPAP puede utilizarse también durante la terapia física para mejorar la separación de esputo.[78] Durante una enfermedad grave, un tubo puede colocarse en la garganta (un procedimiento conocido como un cánula de traqueostomía) para permitir respirar apoyado por un ventilador.
Para los niños que viven con FQ, estudios preliminares muestran pediátrica masaje terapia puede mejorar los pacientes y su calidad de vida de las familias, aunque deben realizarse estudios más rigurosos.[79]
Trasplante
Trasplante de pulmón a menudo se hace necesario para las personas con fibrosis quística como función pulmonar y tolerancia al ejercicio declina. Aunque solo trasplante del pulmón es posible en otras enfermedades, las personas con FQ deben tener ambos pulmones reemplazados porque el pulmón restante podría contener bacterias que pueden infectar el pulmón trasplantado. Un trasplante de páncreas o hígado puede realizarse al mismo tiempo con el fin de aliviar la enfermedad hepática y diabetes.[80] El trasplante de pulmón se considera cuando la función pulmonar disminuye hasta el punto donde se requiere la ayuda de dispositivos mecánicos o supervivencia de los pacientes está amenazada.[81]
Otros aspectos

Los recién nacidos con la obstrucción intestinal generalmente requieren cirugía, mientras que los adultos con distal síndrome de obstrucción intestinal generalmente no. Tratamiento de la insuficiencia pancreática mediante el reemplazo de enzimas digestivas que falta permite el duodeno absorber correctamente los nutrientes y vitaminas que de lo contrario se perdería en las heces.Hasta el momento, ninguna investigación a gran escala que involucra la incidencia de ateroesclerosis y enfermedad cardíaca coronaria en adultos con fibrosis quística se ha realizado. Esto es probablemente debido al hecho de que la inmensa mayoría de las personas con fibrosis quística no vivir lo suficiente para desarrollar ateroesclerosis clínicamente significativa o cardiopatía coronaria.
Diabetes es la complicación más común no pulmonar de CF. It características de mezclas de tipo 1 y tipo 2 diabetes y es reconocida como una entidad distinta, diabetes relacionada con la fibrosis quística (CFRD).[33][82] Tiempo oral medicamentos contra la diabetes a veces se utilizan, se recomienda el único tratamiento es el uso de insulina las inyecciones o un bomba de insulina,[83] y, a diferencia de en la diabetes tipo 1 y 2, las restricciones dietéticas no son recomendables.[33]
Desarrollo de osteoporosis puede prevenirse mediante el aumento de la ingesta de vitamina D y calcioy pueden ser tratados por bifosfonatos, aunque efectos adversos puede ser un problema.[84] Crecimiento deficiente puede evitarse mediante la inserción de un tubo de alimentación para aumentar la calorías a través de alimentos suplementarios o administración de la inyección hormona de crecimiento.[85]
Las infecciones sinusales son tratadas por ciclos prolongados de antibióticos. El desarrollo de los pólipos nasales u otros cambios crónicos dentro de los conductos nasales puede limitar severamente el flujo de aire por la nariz y con el tiempo reducir el olfato del paciente. La cirugía del seno se utiliza a menudo para aliviar la obstrucción nasal y limitar aún más las infecciones. Esteroides nasales tales como fluticasona se utilizan para disminuir la inflamación nasal.[86]
Infertilidad femenina puede ser superada por reproducción asistida tecnología, particularmente transferencia de embriones técnicas. Infertilidad masculina causada por la ausencia de la conducto deferente pueden ser superados con extracción testicular de espermatozoides (TESE), recogida de espermatozoides directamente de los testículos. Si la muestra contiene muy pocas células de la esperma que es probable que un espontáneo fertilización, inyección intracitoplasmática de espermatozoides se puede realizar.[87] Reproducción de terceros es también una posibilidad para las mujeres con FQ.
Pronóstico
El pronóstico para la fibrosis quística ha mejorado debido al diagnóstico precoz a través de la proyección, el mejor tratamiento y acceso a servicios de salud. En 1959, la edad mediana de la supervivencia de los niños con fibrosis quística en Estados Unidos fue de seis meses.[88] En 2010, la supervivencia se estima en 37 años para mujeres y 40 hombres.[4] En Canadá, la mediana de la supervivencia aumentó de 24 años en 1982 a 47,7 en 2007.[89]
De las personas con fibrosis quística que son más de 18 años de edad a partir de 2009, el 92% se había graduado de High School secundaria67% tenían al menos un título universitario, 15% fueron desactivadas y 9% eran desempleado, 56% eran soltero y 39% estaban casados o viviendo con una pareja.[90] En Rusia el total promedio de edad de los pacientes es de 25, que es causada por la ausencia o el alto costo de los medicamentos y el hecho de que no se realiza el trasplante de pulmón.[91]
Calidad de vida
Enfermedades crónicas pueden ser muy difíciles de manejar. Fibrosis quística (FQ) es una enfermedad crónica que afecta a los "digestivos y respiratorios tractos resultando en desnutrición generalizada y las infecciones respiratorias crónicas".[92] Las secreciones gruesas obstruyen las vías respiratorias en los pulmones, que a menudo causan inflamación y las infecciones pulmonares graves.[7][93] Si se está comprometido, que afecta a la calidad de vida de alguien con CF y su capacidad de completar tareas como las tareas cotidianas. Es importante para los pacientes con FQ entender la relación perjudicial que las enfermedades crónicas en la calidad de vida. Según Schmitz y Goldbeck (2006), el hecho de que la fibrosis quística aumenta significativamente la tensión emocional en el individuo y la familia "y la rutina diaria desperdiciador de tiempo necesario de tratamiento puede tener efectos más negativos sobre la calidad de vida (CDV)".[94] Sin embargo, Havermans y colegas (2006) han demostrado que jóvenes pacientes ambulatorios con FQ que han participado en la CFQ-R (Fibrosis quística cuestionario EPQR) "rated algunos dominios de CDV más altos que sus padres".[95] En consecuencia, pacientes con fibrosis quística tienen una actitud más positiva para ellos mismos. Además, hay muchas maneras de mejorar la CdV en pacientes con FQ. Ejercicio es promovido para aumentar la función pulmonar. El hecho de integrar un régimen de ejercicio en la rutina diaria del paciente CF puede mejorar significativamente la calidad de vida.[96] No hay ninguna cura definitiva para la Fibrosis quística. Sin embargo, existen diversos medicamentos que se usan como, mucolíticos, broncodilatadores, esteroides y antibióticos que tienen el propósito de aflojar la mucosidad, ampliar las vías respiratorias, disminuyendo la inflamación y combatir infecciones pulmonares.[97]
Epidemiología
| Mutación | Frecuencia en todo el mundo[98] |
|---|---|
| ΔF508 | 66 – 70%[19] |
| G542X | 2.4% |
| G551D | 1,6% |
| N1303K | 1.3% |
| W1282X | 1.2% |
| Todos los demás | 27,5% |
La fibrosis quística es la más común acortan la vida enfermedad autosómica recesiva entre gente de Europea herencia.[99] En los Estados Unidos, aproximadamente 30.000 individuos tienen CF; la mayoría es diagnosticada por seis meses de edad. En Canadá, existen aproximadamente 4.000 personas con FQ.[100] Aproximadamente 1 de cada 25 personas de ascendencia europea y en 30 de los americanos caucásicos,[101] es un portador de una mutación de fibrosis quística. Aunque es menos común en estos grupos, aproximadamente 1 en 46 CF Hispanos1 en 65 Africanos y 1 de 90 Asiáticos llevar al menos un gen CFTR anormal.[102][103] Irlanda tiene mayor incidencia del mundo de la fibrosis quística, en 1:1353.[104]
Aunque técnicamente un enfermedad rara, la fibrosis quística es catalogada como una de las enfermedades genéticas más extendidas de vida-acortamiento. Es más común entre las Naciones en el mundo occidental. Una excepción es Finlandia, donde sólo uno de cada 80 personas llevan una mutación CF.[105] El Organización Mundial de la salud afirma que "en la Unión Europea, 1 en 2000 – 3000 recién nacidos se encuentra afectada por CF".[6] En los Estados Unidos, 1 en 3.500 niños nacen con fibrosis quística.[106] En 1997, alrededor de 1 en 3.300 niños caucásicos en Estados Unidos nació con fibrosis quística. En contraste, solamente 1 en 15.000 niños afroamericanos sufrieron de fibrosis quística, y en asiático-americanos la tasa fue incluso menor a 1 en 32.000.[107]
La fibrosis quística se diagnostica igualmente en machos y hembras. Por razones que todavía no están claros, datos han demostrado que los varones tienden a tener un más esperanza de vida que las hembras,[108][109] Sin embargo estudios recientes sugieren que esta brecha de género ya no puede existir tal vez debido a las mejoras en las instalaciones sanitarias,[110][111] mientras que un estudio reciente de Irlanda identificó un vínculo entre la hormona femenina estrógeno y peores resultados en la FQ.[112]
La distribución de los alelos CF varía entre poblaciones. La frecuencia de portadores de ΔF508 se ha estimado en 1: 200 en el norte de Suecia, 1:143 de los lituanos, 1:38 en Dinamarca. No ΔF508 los portadores fueron encontrados entre 171 Finlandeses y 151 Pueblo saami.[113] ΔF508 ocurren en Finlandia, pero es un alelo minoría allí. La fibrosis quística es sabida para ocurrir en sólo 20 familias (pedigrees) en Finlandia.[114]
Evolución
El ΔF508 mutación se estima que hasta 52.000 años.[115] Numerosas hipótesis se han adelantado en cuanto a por qué tal mutación letal ha persistido y extendido en la población humana. Otras enfermedades recesivos autosómico comunes tales como anemia falciforme se han encontrado para proteger a los portadores de otras enfermedades, un concepto conocido como ventaja del heterocigoto. Resistencia a los siguientes han sido propuestos como posibles fuentes de ventaja del heterocigoto:
- Cólera: con el descubrimiento que toxina del cólera requiere proteínas CFTR normal host para funcionar correctamente, se planteó la hipótesis que portadores de genes mutantes de CFTR se beneficiaron de resistencia a la cólera y otras causas de diarrea.[116] Otros estudios no han confirmado esta hipótesis.[117][118]
- Fiebre tifoidea: Proteínas CFTR Normal también son esenciales para la entrada de La Salmonella Typhi en las células,[119] sugiriendo que los portadores de genes mutantes de CFTR podrían ser resistentes a fiebre tifoidea. No en vivo estudio ha confirmado esto. En ambos casos, el bajo nivel de fibrosis quística fuera de Europa, en lugares donde la cólera y la fiebre tifoidea fiebre son endémica, no es inmediatamente explicable.
- Diarrea:: Se ha presumido también que la prevalencia de la FQ en Europa podría estar relacionada con el desarrollo de la domesticación del ganado. En esta hipótesis, los portadores de un mutante único cromosoma CFTR tenían cierta protección contra la diarrea causada por intolerancia a la lactosa, antes de la aparición de las mutaciones que creó la tolerancia a la lactosa.[120]
- Tuberculosis:: Otra posible explicación es que los portadores del gen podrían tener cierta resistencia a la tuberculosis.[121][122]
Historia

Se supone que CF apareció alrededor del 3.000 A.C. debido a la migración de los pueblos, las mutaciones genéticas y nuevas condiciones en alimento.[123] Aunque el espectro clínico de la FQ no fue reconocido hasta la década de 1930, ciertos aspectos de la FQ se identificaron mucho antes. De hecho, advirtió literatura procedentes de Alemania y Suiza en el siglo XVIII Wehe dem tipo, das beim Kuß auf die Stirn salzig schmekt, er ist verhext und muss sterbe Calvo o "¡ Ay del niño que sabe salado de un beso en la frente, porque está maldito y pronto debe morir," Reconociendo la asociación entre la enfermedad y la pérdida de la sal en la FQ.[123]
En el siglo XIX, Carl von Rokitansky se describe un caso de muerte fetal con peritonitis del meconium, una complicación del íleo meconial asociada con fibrosis quística. Íleo meconial fue descrita por primera vez en 1905 por Karl Landsteiner.[123] En 1936, Guido Fanconi publicó un artículo que describe una conexión entre enfermedad celíaca, la fibrosis quística del páncreas, y bronquiectasia.[124]
En 1938 Dorothy Hansine Andersen publicó un artículo, "Fibrosis Quística del páncreas y su relación a enfermedad celiaca: un clínico y estudio patológico," en el Diario americano de las enfermedades de los niños. Era el primer para describir la característica la fibrosis quística del páncreas y correlacionarla con el pulmón y la enfermedad intestinal prominente en la FQ.[9] Ella también primero presume que CF es una enfermedad recesiva y utilizó por primera vez reemplazo de enzimas pancreáticas para tratar a los niños afectados. En 1952 Paul di Sant' Agnese descubierto anomalías en sudor electrólitos; un prueba del sudor fue desarrollado y mejorado durante la próxima década.[125]
El primer vínculo entre CF y otro marcador (Paroxonase) fue encontrado en 1985, lo que indica que sólo un locus existe para CF Hans Eiberg. En 1988 la primera mutación de CF, ΔF508 fue descubierto por Francis Collins, Lap-Chee Tsui y John R. Riordan en el cromosoma séptimo. Las investigaciones posteriores ha encontrado más de 1.000 diferentes mutaciones que causan CF.
Debido a mutaciones en el gen CFTR son generalmente pequeñas, genética clásica técnicas habían sido incapaces de localizar con precisión el gen mutado.[126] Uso de marcadores de proteínas, gen-el acoplamiento los estudios fueron capaces de asignar la mutación en el cromosoma 7. Cromosoma-caminando y -saltar luego se utilizaron técnicas para identificar y secuencia el gen.[127] En 1989 Lap-Chee Tsui dirigió un equipo de investigadores de la Hospital para niños enfermos en Toronto descubrió que el gen responsable de la fibrosis quística CF. representa un ejemplo clásico de cómo un trastorno genético humano fue aclarado estrictamente por el proceso de genética hacia adelante.
Investigación
Terapia génica
Terapia génica se ha explorado como una cura potencial para la fibrosis quística. Idealmente, la terapia génica pone una copia normal de la Gen CFTR en las células afectadas. Transferir el gen CFTR normal en las células del epitelio afectado se traduciría en la producción de CFTR funcional en todas las células diana, sin reacciones adversas o una respuesta de inflamación. Los estudios han demostrado que para prevenir las manifestaciones pulmonares de la fibrosis quística, sólo 5 – 10% de la cantidad normal de CFTR expresión génica es necesario.[128] Múltiples enfoques han sido probados para la transferencia de genes, como liposomas y vectores virales en modelos animales y ensayos clínicos. Sin embargo, ambos métodos fueron encontrados para ser las opciones de tratamiento relativamente ineficientes.[129] La razón principal es que muy pocas células toman el vector y expresan el gen, por lo que el tratamiento tiene poco efecto. Además, se han observado problemas en cDNA recombinación, tal que se inutilizó el gen introducido por el tratamiento.[130] Ha habido una reparación funcional en la cultura de CFTR por CRISPR/Cas9 en células madre intestinales organoides de pacientes con fibrosis quística.[131]
Pequeñas moléculas
Un número de pequeñas moléculas que tienen como objetivo compensar las varias mutaciones del gen CFTR se están desarrollando. Un enfoque es desarrollar fármacos que consiguen el ribosoma para superar la codón de parada y sintetizar una proteína CFTR integral. Aproximadamente el 10% de la FQ el resultado de un codón de parada prematuro en el ADN, llevando a la terminación anticipada de la síntesis de proteínas y proteínas truncadas. Objetivo de estas drogas mutaciones de absurdo como G542X, que consiste en el aminoácido Glicina en posición 542 siendo reemplazada por un codón de parada. Antibióticos aminoglucósidos interfieren con la síntesis de proteínas y corrección de errores. En algunos casos, pueden causar la célula superar un codón de parada prematuro mediante la inserción de un aminoácido al azar, permitiendo la expresión de una proteína integral.[132] Los aminoglucósidos gentamicina se ha utilizado para tratar las células pulmonares de pacientes con FQ en el laboratorio para inducir a las células a crecer las proteínas integrales.[133] Otra droga dirigidos a las mutaciones de absurdo es ataluren, que está en ensayos clínicos fase III a partir de octubre de 2011[actualización].[134]
Ivacaftor (Kalydeco), aprobado para su uso por la FDA en los Estados Unidos en enero de 2012[135] objetivos de la mutación G551D)Glicina en posición 551 es sustituido con ácido aspártico). Lumacaftor tiene como objetivo F508del (fenilalanina en la posición 508 está desaparecido).[136]
Sociedad y cultura
- Enfermo: La vida y la muerte de Bob Flanagan, Supermasochist
- 65_Redroses
- Alex: La vida de un niño por Frank Deford
- 3, episodio 1 de la serie Llamar a la partera se refiere a la enfermedad. Es el momento diagnóstico crítico cuando la monja pedante, que ha descubierto que la literatura anterior en su pequeña colección, besos en la frente del bebé y considera el sabor salado para ser concluyente de la condición previamente no identificada pero describe bien.
- Respirar para vivir, un libro de memorias por Laura Rothenberg
Referencias
- ^ Enfermedades de la mutación #Specific causadas por mutaciones de punto punto
- ^ Yankaskas JR, Marshall BC, B Sufian, Simon RH, Rodman D (2004). "Informe de la Conferencia del consenso cuidado adulto fibrosis quística". Pecho 125 (90010): 1 – 39. Doi:10.1378/Chest.125.1_suppl.1s. PMID14734689.
- ^ a b Mishra A, chicharrones R, Massie J (noviembre de 2005). "La relevancia de pruebas para el diagnóstico de la fibrosis quística en la era postgenómica de sudor".. El bioquímico clínico. Comentarios / australiano Asociación de bioquímicos clínicos 26 (4): 135 – 53. PMC1320177. PMID16648884.
- ^ a b MacKenzie, T; Gifford, AH; Sabadosa, KA; Quinton, HB; Knapp, EA; Goss, CH; Marshall, BC (19 de agosto de 2014). "Longevidad de los pacientes con fibrosis quística en el año 2000 a 2010 y más allá: análisis de supervivencia del registro paciente cystic fibrosis foundation.". Anales de medicina interna 161 (4): 233-41. PMID25133359.
- ^ "La fibrosis quística - Mayo Clinic". Mayo Clinic. 30 de noviembre de 2013.
- ^ a b "QUIÉN | Los genes y la enfermedad humana". Who.int. 2010-12-07. 23 / 01 / 2013 obtenido.
- ^ a b Ratjen F, Döring G (febrero de 2003). "Fibrosis quística". The Lancet 361 (9358): 681 – 9. Doi:10.1016/S0140-6736 (03) 12567-6. PMID12606185.
- ^ "La fibrosis quística - National Library of Medicine - PubMed Health". Salud de PubMed. Centro Nacional de información biotecnológica. 30 de noviembre de 2013.
- ^ a b Andersen DH (1938). "La fibrosis quística del páncreas y su relación con la enfermedad celíaca: un estudio clínico y patológico". Am J Dis Child 56:: 344 – 399. Doi:10.1001/archpedi.1938.01980140114013.
- ^ Quinton PM (junio de 2007). "La fibrosis quística: lecciones de la glándula de sudor". Fisiología (Bethesda) 22 (3): 212 – 25. Doi:10.1152/Physiol.00041.2006. PMID17557942.
- ^ a b Hardin DS (agosto de 2004). "GH mejora el crecimiento y el estado clínico en niños con fibrosis quística – una revisión de estudios publicados". EUR j Endocrinol. 151 (Suppl 1): S81 – 5. Doi:10.1530/eje.0.151S081. PMID15339250.
- ^ a b De Lisle RC (septiembre de 2009). "Pasar el bicarbonato: la importancia de HCO3-para el lanzamiento de mucina". J. Clin. Invertir. 119 (9): 2535 – 7. Doi:10.1172/JCI40598. PMC2735941. PMID19726878.
- ^ O ' Malley CA (mayo de 2009). "Control de la infección en la fibrosis quística: cohorte, la contaminación cruzada y el terapeuta respiratorio". Respir Care 54 (5): 641 – 57. Doi:10.4187/aarc0446. PMID19393108.
- ^ Makker K, A, Agarwal Sharma R (abril de 2009). "El estrés oxidativo y la infertilidad masculina". Indio J. med Res. 129 (4): 357 – 67. PMID19535829.
- ^ Blackman SM, Deering-Brose R, R McWilliams, Naughton K, B Coleman, Lai T, Algire M, Beck S, Hoover-Fong J, Hamosh A, Fallin MD, West K, amiento DE, Chakravarti A, Cutler DJ, corte GR (octubre de 2006). "La contribución relativa de los modificadores genéticos y judia a la obstrucción intestinal en la fibrosis quística". Gastroenterología 131 (4): 1030 – 9. Doi:10.1053/j.gastro.2006.07.016. PMC1764617. PMID17030173.
- ^ Ratjen FA (mayo de 2009). "La fibrosis quística: patogenesia y las estrategias terapéuticas futuras". Respir Care 54 (5): 595 – 605. Doi:10.4187/aarc0427. PMID19393104.
- ^ Reaves J, Wallace G (2010). "Inexplicables moretones: pesando los pros y los contras de las posibles causas". Consultor para los pediatras 9:: 201 – 2.
- ^ Canal PA, PJ Mogayzel Jr, Robinson KA, et al (marzo de 2010). "La Fibrosis quística pulmonar directrices: complicaciones pulmonares: hemoptisis y neumotórax". Am J Respir Crit Care Med 182 (3): 298. Doi:10.1164/RCCM.201002-0157OC. PMID20299528.
- ^ a b c d e f g h i j k l m n o p q r s t u v w Mitchell, Richard Sheppard; Kumar, Vinay; Robbins, Stanley L.; Abbas, Abul K.; Fausto, Nelson (2007). Patología básica Robbins. Saunders/Elsevier. ISBN1-4160-2973-7.
- ^ a b c d e f Rowe SM, Miller S, Sorscher EJ (mayo de 2005). "La fibrosis quística". El New England Journal of Medicine 352 (19): 1992-2001. Doi:10.1056/NEJMra043184. PMID15888700.
- ^ Girón RM, Domingo D, B Buendía, Antón E, Ruiz-Velasco LM, Ancochea J (octubre de 2005). "Las micobacterias no tuberculosas en pacientes con fibrosis quística". Arch Bronconeumol. (en Español; Castellano) 41 (10): 560 – 5. Doi:10.1016/S1579-2129 (06) 60283-8. PMID16266669.
- ^ Franco LP, PA Camargos, Becker HM, Guimarães RE (diciembre de 2009). "La evaluación endoscópica nasal de niños y adolescentes con fibrosis quística". Braz J Otorhinolaryngol 75 (6): 806 – 13. Doi:10.1590/S1808-86942009000600006. PMID20209279.
- ^ Maldonado M, Martínez A, Alobid I, Mullol J (diciembre de 2004). "El polyp del antrochoanal". Rinología 42 (4): 178 – 82. PMID15626248.
- ^ Ramsey B, Richardson MA (septiembre de 1992). "Impacto de la sinusitis en la fibrosis quística". J. allergy Clin. Immunol. 90 (3 Pt 2): 547 – 52. Doi:10.1016/0091-6749 (92) 90183-3. PMID1527348.
- ^ Eggermont E, De Boeck K (octubre de 1991). "Anormalidades pequeño-intestinal en pacientes con fibrosis quística". EUR j Pediatr. 150 (12): 824 – 8. Doi:10.1007/BF01954999. PMID1743211.
- ^ Kulczycki LL, Shwachman H (agosto de 1958). "Estudios en la fibrosis quística del páncreas; ocurrencia del prolapso rectal". N. Engl. J. Med. 259 (9): 409 – 12. Doi:10.1056/NEJM195808282590901. PMID13578072.
- ^ Cohn JA, Friedman KJ, Noone PG, Knowles Sr, Silverman LM, Jowell PS (septiembre de 1998). "Relación entre las mutaciones del gen de la fibrosis quística y pancreatitis idiopática". N. Engl. J. Med. 339 (10): 653-8. Doi:10.1056/NEJM199809033391002. PMID9725922.
- ^ Malfroot A, me Dab (noviembre de 1991). "New insights de reflujo gastro-esofágico en la fibrosis quística por seguimiento longitudinal". Arco Dis. niño. 66 (11): 1339 – 45. Doi:10.1136/ADC.66.11.1339. PMC1793275. PMID1755649.
- ^ Khoshoo V, Udall JN (febrero de 1994). "Íleo meconial equivalente en niños y adultos". AM j Gastroenterol. 89 (2): 153 – 7. PMID8304294.
- ^ Williams SG, Mowat Westaby D, MS Tanner, AP (octubre de 1992). Hígado y problemas biliares en la fibrosis quística. Br. med Bull. 48 (4): 877 – 92. PMID1458306.
- ^ Colombo C, Russo MC, Zazzeron L, Romano G (julio de 2006). "Enfermedad del hígado en la fibrosis quística". J Pediatr. Gastroenterol. NUTR. 43 (Suppl 1): S49 – 55. Doi:10.1097/01.mpg.0000226390.02355.52. PMID16819402.
- ^ Moran A, Pyzdrowski KL, Weinreb J, Kahn BB, Smith SA, KS Adams, Seaquist ER (agosto de 1994). "Sensibilidad a la insulina en la fibrosis quística". Diabetes 43 (8): 1020 – 6. Doi:10.2337/diabetes.43.8.1020. PMID8039595.
- ^ a b c Alves Cde A, RA, Aguiar Alves AC, Santana MA (abril de 2007). "Diabetes mellitus en pacientes con fibrosis quística". J Bras Pneumol 33 (2): 213 – 21. Doi:10.1590/S1806-37132007000200017. PMID17724542.
- ^ Haworth CS, Selby PL, Webb AK, ME Dodd, Musson H, McL Niven R, G Economou, Horrocks AW, Freemont AJ, Mawer EB, Adams JE (noviembre de 1999). "Baja densidad mineral ósea en adultos con fibrosis quística". Tórax 54 (11): 961 – 7. Doi:10.1136/THX.54.11.961. PMC1745400. PMID10525552.
- ^ Vandemergel X, G Decaux (abril de 2003). «[Revisión de Osteoartropatía hipertrófica y aporrear digital]». Revista Médicale de Bruxelles (en francés) 24 (2): 88-94. PMID12806875.
- ^ Pitts-Tucker TJ, Miller MG, Littlewood JM (junio de 1986). "Aporrear del dedo en la fibrosis quística". Arco Dis. niño. 61 (6): 576 – 9. Doi:10.1136/ADC.61.6.576. PMC1777828. PMID3488032.
- ^ McCallum TJ, Milunsky JM, Cunningham DL, Harris DH, Maher TA, Oates RD (octubre de 2000). "La fertilidad en hombres con fibrosis quística: una actualización sobre las actuales prácticas quirúrgicas y los resultados". Pecho 118 (4): 1059 – 62. Doi:10.1378/Chest.118.4.1059. PMID11035677.
- ^ Chen H, Ruan YC, Xu WM, Chen J, Chan HC (2012). "Regulación de la fertilidad masculina por CFTR y las implicaciones en la infertilidad masculina". Actualización de la reproducción humana 18 (6): 703-713. Doi:10.1093/humupd/dms027. PMID22709980.
- ^ Augarten A, Yahav Y, Kerem BS, Halle D, Laufer J, Szeinberg A, Dor J, S Mashiach, Gazit E, Madgar I (noviembre de 1994). "Ausencia bilateral congénita de conductos deferentes en la ausencia de fibrosis quística". The Lancet 344 (8935): 1473 – 4. Doi:10.1016/S0140-6736 (94) 90292-5. PMID7968122.
- ^ M Gilljam, M Antoniou, Shin J, Dupuis A, Corey M, DE Tullis (julio de 2000). "El embarazo en la fibrosis quística. Resultado fetal y materno". Pecho 118 (1): 85-91. Doi:10.1378/Chest.118.1.85. PMID10893364.
- ^ "Perfil: Lap-Chee Tsui". Science.ca. 1989-05-09. 23 / 01 / 2013 obtenido.
- ^ Bobadilla JL, Macek M, bien JP, Farrell PM (junio de 2002). "La fibrosis quística: un análisis de las mutaciones de CFTR en todo el mundo — correlación con los datos de incidencia y aplicación a la investigación". Zumbido. Mutat. 19 (6): 575 – 606. Doi:10.1002/humu.10041. PMID12007216.
- ^ DB corto, Trotter KW, Reczek D, Kreda SM, Bretscher A, Boucher RC, Stutts MJ, Milgram SL (julio de 1998). "Una proteína PDZ apical anclas el regulador de conductancia transmembrana de la fibrosis quística al citoesqueleto". J Biol Chem. 273 (31): 19797 – 801. Doi:10.1074/JBC.273.31.19797. PMID9677412.
- ^ Childers, Eckel & Himmel 2007
- ^ Xu, Szép & Lu 2009
- ^ Moskwa, Lorentzen & Excoffon 2007
- ^ Conner, Wijkstrom-Frei & Randell 2007
- ^ a b Saiman L (2004). "Microbiología de la enfermedad pulmonar CF temprana". Comentarios respiratoria pediátrica 5 (Suppl A): S367 – 69. Doi:10.1016/S1526-0542 (04) 90065-6. PMID14980298.
- ^ Tümmler B, Koopmann U, Grothues D, H Weissbrodt, Steinkamp G, von der Hardt H (junio de 1991). "Adquisición Nosocomial de Pseudomonas aeruginosa en pacientes con fibrosis quística". J. Clin. Latinoamericana de investigación pediátrica. 29 (6): 1265 – 7. Bibcode:1991JPoSA...29.1265A. Doi:10.1002/Pola.1991.080290905. PMC271975. PMID1907611.
- ^ "Pseudomonas cepacia en campamentos de verano para las personas con fibrosis quística". MMWR Morb. Mortal. Wkly. Rep. 42 (23): 456 – 9. Junio de 1993. PMID7684813.
- ^ Pegues DA, LA Carson, Tablan OC, SC FitzSimmons, Roman SB, Miller JM, Jarvis WR (mayo de 1994). "Adquisición de Pseudomonas cepacia en campamentos de verano para los pacientes con fibrosis quística. Verano Campamento de grupo de estudio". J Pediatr. 124 (5 Pt 1): 694 – 702. Doi:10.1016/S0022-3476 (05) 81357-5. PMID7513755.
- ^ Pankhurst CL, Philpott-Howard J (abril de 1996). "Los factores de riesgo ambientales asociados con el equipo médico y dental en la transmisión de cepacia de Burkholderia (Pseudomonas) en pacientes con fibrosis quística". J. hospital infectar. 32 (4): 249-55. Doi:10.1016/S0195-6701 (96) 90035-3. PMID8744509.
- ^ Jones AM, Govan JR, Doherty CJ, ME Dodd, Isalska BJ, Stanbridge TN, Webb AK (junio de 2003). "Identificación de difusión en el aire de epidemias cepas multirresistentes de Pseudomonas aeruginosa en un centro de CF durante un brote de infección cruzada". Tórax 58 (6): 525 – 27. Doi:10.1136/Thorax.58.6.525. PMC1746694. PMID12775867.
- ^ Høiby N (junio de 1995). Aislamiento y tratamiento de pacientes con fibrosis quística con infecciones pulmonares causadas por Pseudomonas (Burkholderia) cepacia y Pseudomonas aeruginosa multirresistentes. Neth J Med 46 (6): 280 – 87. Doi:10.1016/0300-2977 (95) 00020-N. PMID7643943.
- ^ a b Pihet M, Carrere J, Cimon B, D Chabasse, Delhaes L, F Symoens, Bouchara JP (junio de 2009). "Ocurrencia y la pertinencia de hongos filamentosos en las secreciones respiratorias de los pacientes con fibrosis quística — una revisión". Med Mycol. 47 (4): 387-97. Doi:10.1080/13693780802609604. PMID19107638.
- ^ Rapaka RR, Kolls JK (2009). "Patogenesia de la aspergilosis broncopulmonar alérgica en la fibrosis quística: conocimiento actual y futuro". Med Mycol. 47 (Suppl 1): S331 – 7. Doi:10.1080/13693780802266777. PMID18668399.
- ^ a b Davies JC, Alton EW, Bush (diciembre de 2007). "Fibrosis quística". BMJ 335 (7632): 1255 – 9. Doi:10.1136/bmj.39391.713229.ad. PMC2137053. PMID18079549.
- ^ Ross LF (septiembre de 2008). "Recién nacido de detección para la fibrosis quística: una lección de disparidades en la salud pública". La revista de Pediatría 153 (3): 308 – 13. Doi:10.1016/j.jpeds.2008.04.061. PMC2569148. PMID18718257.
- ^ Assael BM, C Castellani, Ocampo MB, Iansa P, Callegaro A, Valsecchi MG (septiembre de 2002). "Análisis de Epidemiología y la supervivencia de la fibrosis quística en un área de cribado neonatal intensa más de 30 años". American Journal of Epidemiology 156 (5): 397-401. Doi:10.1093/AJE/kwf064. PMID12196308.
- ^ Minarowski, arenas & Minarowska 2008
- ^ Popa RC (febrero de 1997). "El diagnóstico de la fibrosis quística". N. Engl. J. Med. 336 (7): 487 – 91. Doi:10.1056/NEJM199702133360707. PMID9017943.
- ^ Freudenheim, Milt (2009-12-22). "Herramienta en la lucha de la Fibrosis quística: un registro". New York Times. págs. D1. 2009-12-21.
- ^ Colegio Americano de Obstetras y ginecólogos y American College of Medical Genetics. Preconcepcional y prenatal portador de detección para la fibrosis quística. Pautas clínicas y del laboratorio. American College of Obstetricians and Gynecologists, Washington, DC, octubre de 2001.
- ^ Elias S, Annas GJ, Simpson JL (abril de 1991). "Portador de detección para la fibrosis quística: implicaciones para la práctica obstétrica y ginecológica". Soy J. Obstet. Gynecol. 164 (4): 1077 – 83. Doi:10.1016/0002-9378 (91) 90589-j. PMID2014829.
- ^ Tabor A, Philip J, M Madsen, Bang J, Obel EB, Nørgaard-Pedersen B (junio de 1986). "Ensayo controlado aleatorio de amniocentesis genética en 4606 mujeres de bajo riesgo". The Lancet 1 (8493): 1287 – 93. Doi:10.1016/S0140-6736 (86) 91218-3. PMID2423826.
- ^ Eddleman KA, Malone FD, Sullivan L, duques K, Berkowitz RL, Kharbutli Y, Porter TF, Luthy DA, Comstock CH, Saade GR, Klugman S, Dugoff L, Craigo SD, Timor-Polka Tritsch IE, Carr SR, Wolfe HM, D'Alton ME (noviembre de 2006). "Tasas de pérdida del embarazo después de la amniocentesis del midtrimester". Obstetrics and Gynecology 108 (5): 1067 – 72. Doi:10.1097/01.AOG.0000240135.13594.07. PMID17077226.
- ^ Davis LB, campeón SJ, justo entonces, Baker VL, Garber AM (abril de 2010). "Un análisis costo / beneficio de diagnóstico genético preimplantacional para parejas de portador de fibrosis quística". Fertil. Esterol. 93 (6): 1793 – 804. Doi:10.1016/j.fertnstert.2008.12.053. PMID19439290.
- ^ Pai VB, Nahata MC (octubre de 2001). Eficacia y seguridad de tobramicina aerosolizada en la fibrosis quística. Pediatr. Pulmonol. 32 (4): 314 – 27. Doi:10.1002/PPUL.1125. PMID11568993.
- ^ Westerman EM Le Brun PP, Touw DJ, Frijlink HW, Heijerman HG (marzo de 2004). "Efecto del sulfato de Colistina nebulizada y colistina sulphomethate sobre la función pulmonar en pacientes con fibrosis quística: un estudio piloto". J. quiste. Fibros. 3 (1): 23 – 8. Doi:10.1016/j.JCF.2003.12.005. PMID15463883.
- ^ McCoy KS AL Quittner, Oermann CM, Gibson RL, GZ Retsch-Bogart, Montgomery AB (noviembre de 2008). "Inhala aztreonam lisina para aeruginosa de los Pseudomonas crónica de las vías respiratorias en la fibrosis quística". AM j Respir. Crit. Care Med. 178 (9): 921 – 8. Doi:10.1164/RCCM.200712-1804OC. PMC2577727. PMID18658109.
- ^ Ryan G, Singh M, Dwan K (2011). "Antibióticos inhalados para el tratamiento a largo plazo en la fibrosis quística". La base de datos Cochrane de revisiones sistemáticas (3): CD001021. Doi:10.1002/14651858.CD001021.pub2. PMID21412868.
- ^ Hansen CR, T Pressler, Koch C, Høiby N (marzo de 2005). "A largo plazo azitromycin el tratamiento de pacientes con fibrosis quística con infección crónica de Pseudomonas aeruginosa; un estudio de cohortes observacionales". J. quiste. Fibros. 4 (1): 35 – 40. Doi:10.1016/j.JCF.2004.09.001. PMID15752679.
- ^ Tan KH, Mulheran M, Knox AJ, Smyth AR (marzo de 2003). "Prescripción de aminoglucósidos y vigilancia en la fibrosis quística". AM j Respir. Crit. Care Med. 167 (6): 819 – 23. Doi:10.1164/RCCM.200109-012CC. PMID12623858.
- ^ van der Schans C, A Prasad, principal E (2000). "La fisioterapia torácica comparada con ninguna fisioterapia torácica para la fibrosis quística". En Van Der Schans, Cees P. Cochrane Database Syst Rev (2): CD001401. Doi:10.1002/14651858.CD001401. PMID10796781.
- ^ Kuver R, Lee SP (abril de 2006). "Solución salina hipertónica para la fibrosis quística". N. Engl. J. Med. 354 (17): 1848-51; Autor respuesta 1848 – 51. Doi:10.1056/NEJMc060351. PMID16642591.
- ^ Lieberman J (julio de 1968). "Efecto de aerosol Dornasa sobre viscosidad de esputo en los casos de fibrosis quística". JAMA 205 (5): 312 – 3. Doi:10.1001/Jama.205.5.312. PMID5694947.
- ^ D Kellerman, Rossi Mospan A, Engels J, Schaberg A, Gorden J, Smiley L (2008). "Denufosol: una revisión de estudios con agonistas inhalados de P2Y(2) que llevó a la fase 3". Terapéutica y Farmacología pulmonar 21 (4): 600 – 7. Doi:10.1016/j.pupt.2007.12.003. PMID18276176.
- ^ Moran F, Bradley JM, Piper AJ (2009). "La ventilación no invasiva para la fibrosis quística". En Moran, Fidelma. Cochrane Database Syst Rev (1): CD002769. Doi:10.1002/14651858.CD002769.pub3. PMID19160211.
- ^ Huth MM, Zink KA, Van Horn NR (2005). "Los efectos de la terapia de masaje en la mejora de los resultados para los jóvenes con fibrosis quística: una revisión de la evidencia". Journal of Pediatric enfe 31 (4): 328 – 32. PMID16229132.
- ^ Fridell JA, Vianna R, Kwo PY, Howenstine M, Sannuti A, Molleston JP, Pescovitz MD, Tector AJ (octubre de 2005). "Simultáneo del hígado y páncreas trasplante en pacientes con fibrosis quística". Trasplante. Proc. 37 (8): 3567 – 9. Doi:10.1016/j.transproceed.2005.09.091. PMID16298663.
- ^ Belkin RA, Henig NR, cantante LG, Chaparro C, Rubenstein RC, Xie SX, Yee JY, Kotloff RM, Lipson DA, Bunin GR (marzo de 2006). "Factores de riesgo para la muerte de los pacientes con fibrosis quística a la espera de trasplante de pulmón". AM j Respir. Crit. Care Med. 173 (6): 659 – 66. Doi:10.1164/RCCM.200410-1369OC. PMC2662949. PMID16387803.
- ^ Zirbes J, Milla CE (septiembre de 2009). "La fibrosis quística relacionada con diabetes". Paediatr Respir Rev 10 (3): 118 – 23; el concurso 123. Doi:10.1016/j.prrv.2009.04.004. PMID19651382.
- ^ Onady GM, Stolfi A (2005). "Insulina y agentes orales para la gestión de la diabetes relacionada con la fibrosis quística". En Onady, Gary M. Cochrane Database Syst Rev (3): CD004730. Doi:10.1002/14651858.CD004730.pub2. PMID16034943.
- ^ Conwell LS, Chang AB (2012). "Los bifosfonatos para la osteoporosis en personas con fibrosis quística". En Conwell, Louise S. Cochrane Database Syst Rev 4 (4): CD002010. Doi:10.1002/14651858.CD002010.pub3. PMID22513903.
- ^ Hardin DS, arroz J, Ahn C, Ferkol T, Howenstine M, Spears S, Prestidge C, Seilheimer DK, pastor R (marzo de 2005). "Tratamiento hormonal de crecimiento mejora la nutrición y el crecimiento en niños con fibrosis quística recibir nutrición enteral". J Pediatr. 146 (3): 324 – 8. Doi:10.1016/j.jpeds.2004.10.037. PMID15756212.
- ^ Marca SC, Kissner DG (1997). "Gestión de la sinusitis en la fibrosis quística para adultos". Soy Rhinol J 11 (1): 11 – 4. Doi:10.2500/105065897781446810. PMID9065342.
- ^ Phillipson GT, Petrucco OM, Matthews CD (febrero de 2000). "Ausencia bilateral congénita de conductos deferentes, análisis de la mutación de fibrosis quística e inyección intracitoplasmática de espermatozoides". Zumbido. Reprod. 15 (2): 431 – 5. Doi:10.1093/humrep/15.2.431. PMID10655317.
- ^ "¿Cuál es la esperanza de vida para las personas que tienen CF (en los Estados Unidos)".. Fundación de Fibrosis quística. 2008. 2010-03-14.
- ^ "Informe de registro datos del paciente canadiense de Fibrosis quística" (PDF). Canadian Cystic Fibrosis Foundation. 2007. 2010-03-14.
- ^ "Informe de datos anuales de registro de enfermos de Fibrosis quística 2009" (PDF). Fundación de Fibrosis quística. 2009.
- ^ "ПЕЙНМЯРПСЙЖХЪ". 31 / 05 / 2012 Mucoviscidos.ru.. 23 / 01 / 2013 obtenido.
- ^ Yu H, Nasr SZ, Deretic V (abril de 2000). "Las defensas pulmonares innata y comprometido Pseudomonas aeruginosa separación en el modelo murino desnutridos de infecciones respiratorias en la fibrosis quística". Infectar. Immun. 68 (4): 2142 – 7. Doi:10.1128/IAI.68.4.2142-2147.2000. PMC97396. PMID10722612.
- ^ Rosenstein BJ, PL Zeitlin (enero de 1998). "Fibrosis quística". The Lancet 351 (9098): 277-82. Doi:10.1016/S0140-6736 (97) 09174-5. PMID9457113.
- ^ Schmitz TG, Goldbeck L (2006). "El efecto de la rehabilitación de pacientes hospitalizados de programas sobre la calidad de vida en pacientes con fibrosis quística: un estudio multicéntrico". Resultados de Qual vida salud 4:: 8. Doi:10.1186/1477-7525-4-8. PMC1373610. PMID16457728.
- ^ Watter Hegarty M, Macdonald J, P, C Wilson (julio de 2009). "Calidad de vida de la gente joven con fibrosis quística: efectos de la hospitalización, la edad y género y las diferencias en las percepciones de padre-hijo". Child Care Health Dev 35 (4): 462 – 8. Doi:10.1111/j.1365-2214.2008.00900.x. PMID18991968.
T Havermans, Vreys M, Proesmans M, De Boeck C (enero de 2006). "Evaluación del acuerdo entre padres e hijos sobre la calidad de vida relacionada con la salud en niños con fibrosis quística". Child Care Health Dev 32 (1): 1 – 7. Doi:10.1111/j.1365-2214.2006.00564.x. PMID16398786. - ^ Moorcroft AJ, ME Dodd, Webb AK (1998). Limitaciones de ejercicio y entrenamiento para los pacientes con fibrosis quística. Favor Egido 20 (6 – 7): 247 – 53. Doi:10.3109/09638289809166735. PMID9637933.
- ^ Canadá de la Fibrosis quística. (2011). los medicamentos. (Catálogo en línea Nº 10684-5100 RR0001.). Obtenido de https://www.cysticfibrosis.CA/en/Treatment/medications.php
- ^ Araújo FG, FC Novaes, Santos NP, Martins VC, Souza SM, Santos SE, Ribeiro-dos-Santos AK (enero de 2005). "Prevalencia de deltaF508, las mutaciones G551D y G542X, R553X entre pacientes con fibrosis quística en el norte de Brasil". Braz. J. med Biol. Res. 38 (1): 11-5. Doi:10.1590/S0100 - 879 X 2005000100003. PMID15665983.
- ^ Tobias, Edward (2011). Genética médica esencial. John Wiley & Sons. p. 312. ISBN1-118-29370-3.
- ^ https://www.cysticfibrosis.CA/en/aboutCysticFibrosis/CfStatistics.php El canadienses hechos y cifras sobre la Fibrosis quística
- ^ Cystic Fibrosis Foundation – prueba genética del portador Actualizado 09/07/07
- ^ Rosenstein BJ, corte GR (abril de 1998). "El diagnóstico de la fibrosis quística: una declaración de consenso. Cystic Fibrosis Foundation consenso Panel". J Pediatr. 132 (4): 589 – 95. Doi:10.1016/S0022-3476 (98) 70344-0. PMID9580754.
- ^ Hamosh A, SC FitzSimmons, Macek M, Knowles Sr, Rosenstein BJ, corte GR (febrero de 1998). "Comparación de las manifestaciones clínicas de la fibrosis quística en pacientes blancos y negros". J Pediatr. 132 (2): 255 – 9. Doi:10.1016/S0022-3476 (98) 70441-X. PMID9506637.
- ^ Farrell P, S Joffe, Foley L, GJ Canny, Mayne P, Rosenberg M (septiembre de 2007). "Diagnóstico de la fibrosis quística en la República de Irlanda: Epidemiología y costos". Ir Med J 100 (8): 557 – 60. PMID17955689.
- ^ Hytönen M, Patjas M, Vento SI, P Kauppi, Malmberg H, Ylikoski J, J Kere (diciembre de 2001). Gen de la fibrosis quística mutaciones deltaF508 y 394delTT en pacientes con sinusitis crónica en Finlandia. Acta Otolaryngol. 121 (8): 945 – 7. Doi:10.1080/000164801317166835. PMID11813900.
- ^ Russell, Peter (2011). Biología: la ciencia dinámica. (2ª ed.). Belmont, CA: Arroyos/Cole, Cengage Learning. p. 304. ISBN978-0-538-49372-7.
- ^ Pruebas genéticas para la fibrosis quística pruebas genéticas para la Fibrosis quística. Institutos nacionales de salud, declaración de conferencia de desarrollo de consenso. 14 – 16 de abril de 1997. Recuperado encendido 20 de noviembre de 2009.
- ^ Rosenfeld M, Davis R, FitzSimmons S, Pepe M, Ramsey B (mayo de 1997). "Brecha de género en la mortalidad de la fibrosis quística". AM j Epidemiol. 145 (9): 794-803. Doi:10.1093/oxfordjournals.AJE.a009172. PMID9143209.
- ^ Coakley RD, Sun H LA Clunes, Rasmussen JE, Stackhouse JR, Okada SF, Fricks yo, joven SL, Fabius R (diciembre de 2008). "17beta-Estradiol inhibe Ca2 +-dependiente homeostasis del volumen líquido superficial de las vías respiratorias en epitelios de las vías respiratorias humanas fibrosis quística". J. Clin. Invertir. 118 (12): 4025 – 35. Doi:10.1172/JCI33893. PMC2582929. PMID19033671.
- ^ Verma N, Bush A, Buchdahl R (octubre de 2005). "Hay todavía una brecha de género en la fibrosis quística?". Pecho 128 (4): 2824 – 34. Doi:10.1378/Chest.128.4.2824. PMID16236961.
- ^ Morán A, J Dunitz, Nathan B, Saeed A, B Holme, Thomas W (septiembre de 2009). "La diabetes relacionada con la fibrosis quística: tendencias en la prevalencia, incidencia y mortalidad". Cuidado de la diabetes 32 (9): 1626 – 31. Doi:10.2337/dc09-0586. PMC2732133. PMID19542209.
- ^ "Peor CF para mujeres ' debido al efecto del estrógeno'". El Irish Times. 08 de agosto de 2010.
- ^ Wennberg C, Kucinskas V (1994). "Baja frecuencia del delta F508 mutación en poblaciones ugrofinesas y báltico". Zumbido. Hered. 44 (3): 169-71. Doi:10.1159/000154210. PMID8039801.
- ^ Kere J, Savilahti E, Norio R, X Estivill, de la Chapelle A (septiembre de 1990). "La fibrosis quística mutación delta F508 Finlandia: predominan otras mutaciones". Zumbido. Genet. 85 (4): 413-5. Doi:10.1007/BF02428286. PMID2210753.
- ^ Wiuf C (agosto de 2001). ¿Heterocigotos delta F508 tiene una ventaja selectiva?". Genet. Res. 78 (1): 41-7. Doi:10.1017/S0016672301005195. PMID11556136.
- ^ Gabriel SE, Brigman KN, Koller BH, Boucher RC, Stutts MJ (octubre de 1994). "Resistencia de heterocigoto de fibrosis quística a la toxina del cólera en el modelo murino de fibrosis quística". Ciencia 266 (5182): 107 – 9. Bibcode:1994Sci...266..107G. Doi:10.1126/science.7524148. PMID7524148.
- ^ Cuthbert AW, J Halstead, R Ratcliff, Colledge WH, Evans MJ (enero de 1995). "La hipótesis de ventaja genética en heterocigotos de la fibrosis quística: un estudio murino". J Physiol (Lond.) 482 (Pt 2): 449-54. PMC1157742. PMID7714835.
- ^ Högenauer C, Santa Ana CA, Porter JL, Millard M, Gelfand A, Rosenblatt RL, Prestidge CB, Fordtran JS (diciembre de 2000). "La secreción de cloruro intestinal activa en humanos portadores de mutaciones de fibrosis quística: una evaluación de la hipótesis de que los heterocigotos tienen secreción subnormal cloruro intestinal activa". Soy J. Hum. Genet. 67 (6): 1422 – 7. Doi:10.1086/316911. PMC1287919. PMID11055897.
- ^ Muelle GB, lechada M, Zaidi T, Meluleni G, Mueschenborn SS, Banting G, R Ratcliff, Evans MJ, Colledge WH (mayo de 1998). "Salmonella typhi utiliza CFTR para entrar en las células epiteliales intestinales". Naturaleza 393 (6680): 79-82. Bibcode:1998Natur.393...49S. Doi:10.1038/30006. PMID9590693.
- ^ Modiano G, BM Ciminelli, Pignatti PF (marzo de 2007). "La fibrosis quística y la persistencia de lactasa: una posible correlación". EUR j Hum. Genet. 15 (3): 255 – 9. Doi:10.1038/sj.ejhg.5201749. PMID17180122.
- ^ Poolman EM, Galvani AP (febrero de 2007). "Evaluando candidatos agentes de presión selectiva para la fibrosis quística". Journal of the Royal Society, interfaz 4 (12): 91 – 8. Doi:10.1098/RSIF.2006.0154. PMC2358959. PMID17015291.
- ^ Williams, N (2006). "Teme la huella por nueva amenaza TB". Current Biology 16 (19): R821. Doi:10.1016/j.Cub.2006.09.009.
- ^ a b c Busch R (1990). "En la historia de la fibrosis quística". Acta Univ Carol Med (Praha) 36 (1 – 4): 13 – 5. PMID2130674.
- ^ Fanconi, G., Uehlinger, E., Knauer, C. (1936). "Das coeliakiesyndrom bei angeborener zysticher pankreasfibromatose und bronchiektasien". Wien. Wschr med. 86:: 753 – 6.
- ^ Agnese di PA, querida RC, Perera GA, Shea E (noviembre de 1953). "Composición del electrólito anormal de sudor en la fibrosis quística del páncreas; significación clínica y relación con la enfermedad". Pediatría 12 (5): 549 – 63. PMID13111855.
- ^ Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL (septiembre de 1989). "Identificación del gen de la fibrosis quística: clonación y caracterización de ADN complementario". Ciencia 245 (4922): 1066 – 73. Bibcode:1989Sci...245.1066R. Doi:10.1126/science.2475911. PMID2475911.
- ^ Rommens JM, Iannuzzi MC, Kerem B, Drumm ML, Melmer G, Dean M, Rozmahel R, Cole JL, Kennedy D, N Hidaka (septiembre de 1989). "Identificación del gen de la fibrosis quística: cromosoma caminando y saltando". Ciencia 245 (4922): 1059 – 65. Bibcode:1989Sci...245.1059R. Doi:10.1126/science.2772657. PMID2772657.
- ^ Meyer Ramalho AS, Beck S, M, D Penque, corte GR, Amaral MD (noviembre de 2002). "Cinco por ciento del regulador de la conductancia transmembrana de fibrosis quística normal mRNA mejora la severidad de la enfermedad pulmonar en la fibrosis quística". AM j Respir. Célula análizar Biol. 27 (5): 619 – 27. Doi:10.1165/RCMB.2001-0004oc. PMID12397022.
- ^ Tate S, Elborn S (marzo de 2005). "El progreso hacia la terapia génica para la fibrosis quística". Expert Opin Drug Deliv 2 (2): 269 – 80. Doi:10.1517/17425247.2.2.269. PMID16296753.
- ^ En línea 'herencia mendeliana en el hombre' (OMIM) FIBROSIS QUÍSTICA; CF-219700
- ^ Schwank G, Koo BK, Sasselli V, JF Dekkers, Heo Demircan T, Sasaki N, S, E Cuppen, Boymans van der Ent CK, Nieuwenhuis EE, Beekman JM, Clevers H (05 de diciembre de 2013). "Reparación funcional de CFTR por CRISPR/Cas9 en células madre intestinales organoides de pacientes con fibrosis quística". Célula de vástago de la célula 13 (6): 653-8. Doi:10.1016/j.Stem.2013.11.002. PMID24315439.
- ^ Dietz HC (agosto de 2010). "Nuevos enfoques terapéuticos para desórdenes mendelianos". N. Engl. J. Med. 363 (9): 852 – 63. Doi:10.1056/NEJMra0907180. PMID20818846. Texto completo gratis
- ^ Wilschanski M, Yahav Y, Yaacov Y, H Blau, Bentur L, Rivlin J, M Aviram, Bdolah-Abram T, Bebok Z, Shushi L, Kerem B, Kerem E (octubre de 2003). "Corrección de gentamicina-inducido de la función CFTR en pacientes con fibrosis quística y CFTR detener las mutaciones". N. Engl. J. Med. 349 (15): 1433 – 41. Doi:10.1056/NEJMoa022170. PMID14534336.
- ^ ClinicalTrials.gov NCT00803205 Estudio de Ataluren (PTC124 ™) en la Fibrosis quística
- ^ "'"BusinessWeek''". BusinessWeek.com. 2012-02-02. 23 / 01 / 2013 obtenido.
- ^ Merk; Schubert-Zsilavecz. "Neue Ansätze bei Mukoviszidose". Pharmazeutische Zeitung (en alemán) 156 (37): 24 – 27.
Lectura adicional
- Etiología fúngica en las infecciones asociadas a CF revisado exhaustivamente por Pihet et al.: Pihet M, Carrere J, Cimon B, D Chabasse, Delhaes L, F Symoens, Bouchara JP (junio de 2009). "Ocurrencia y la pertinencia de hongos filamentosos en las secreciones respiratorias de los pacientes con fibrosis quística — una revisión". Med Mycol. 47 (4): 387-97. Doi:10.1080/13693780802609604. PMID19107638.
- Moskwa P, D Lorentzen, Excoffon KJ, Zabner J, McCray PB, Nauseef WM, Dupuy C, Bánfi B (enero de 2007). "Un sistema de defensa anfitrión novela de vías respiratorias es defectuoso en la fibrosis quística". AM j Respir. Crit. Care Med. 175 (2): 174 – 83. Doi:10.1164/RCCM.200607-1029OC. PMC2720149. PMID17082494.
- Childers M, G Eckel, Himmel A, Caldwell J (2007). "Un nuevo modelo de patología de la fibrosis quística: falta de transporte de glutatión y sus conjugados de tiocianato". Hipótesis médicas 68 (1): 101 – 12. Doi:10.1016/j.mehy.2006.06.020. PMID16934416.
- Conner GE, Salathe M, Forteza R (diciembre de 2002). "Metabolismo lactoperoxidasa y peróxido de hidrógeno en la vía aérea". AM j Respir. Crit. Care Med. 166 (12 Pt 2): S57 – 61. Doi:10.1164/RCCM.2206018. PMID12471090.
- Conner Randell de GE, Wijkstrom-Frei C, SH, Fernandez View, Salathe M (enero de 2007). "El sistema lactoperoxidasa enlaces de transporte de anión a la defensa del huésped en la fibrosis quística". FEBS Letters 581 (2): 271 – 78. Doi:10.1016/j.febslet.2006.12.025. PMC1851694. PMID17204267.
- Minarowski Ł, arenas D, Minarowska A, Karwowska A, Sulewska A, Gacko M, Chyczewska E (2008). "Concentración de tiocianato en la saliva de pacientes con fibrosis quística". Folia neuroblasto. Cytobiol. 46 (2): 245 – 6. Doi:10.2478/v10042-008-0037-0. PMID18519245.
- Rada B, Leto TL (2009). "Guerra de redox entre las células epiteliales de las vías respiratorias y Pseudomonas: dual oxidasa versus pyocyanin". Immunol. Res. 43 (1 – 3): 198 – 209. Doi:10.1007/s12026-008-8071-8. PMC2776630. PMID18979077.
- Fischer H (octubre de 2009). "Los mecanismos y la función de DUOX en epitelios del pulmón". Antioxid. Señal de redox. 11 (10): 2453 – 65. Doi:10.1089/ARS.2009.2558. PMC2823369. PMID19358684.
- Pedemonte N, Caci E, E Sondo, Caputo A, K Rhoden, Pfeffer U, Di Candia M, Bandettini R, R Ravazzolo, Zegarra-Moran O, Galietta LJ (abril de 2007). "Transporte de tiocianato en reposo y células epiteliales bronquiales humanas IL-4-estimulados: papel de los canales pendrin y anión". J. Immunol. 178 (8): 5144 – 53. Doi:10.4049/jimmunol.178.8.5144. PMID17404297.
- C Wijkstrom-Frei, El-Chemaly S, R Ali-Rachedi, Gerson C, MA Cobas, Forteza R, Salathe M, Conner GE (agosto de 2003). "Lactoperoxidasa y defensa del huésped humano vía aérea". AM j Respir. Célula análizar Biol. 29 (2): 206 – 12. Doi:10.1165/RCMB.2002-0152OC. PMID12626341.
- Xu Y, Szép S, Lu Z (diciembre de 2009). "El papel antioxidante de tiocianato en la patogenia de la fibrosis quística y otras enfermedades relacionadas con inflamación". Proc. nacional Acad. Sci U.S.A. 106 (48): 20515 – 9. Bibcode:2009PNAS...10620515 X. Doi:10.1073/pnas.0911412106. PMC2777967. PMID19918082.
Enlaces externos
| Wikimedia Commons tiene medios relacionados con Fibrosis quística. |
- Fibrosis quística en DMOZ
- cf en NIH/UW GeneTests
- CF-Europa.UE
- GeneCards búsqueda de genes involucrados en la Fibrosis quística
- Base de datos mutación de Fibrosis quística
- Extracto de herencia mendeliana del hombre en línea de la Fibrosis quística
|
||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||